Convolutional Networks on Graphs for Learning Molecular Fingerprints
We introduce a convolutional neural network that operates directly on graphs. These networks allow end-to-end learning of prediction pipelines whose inputs are graphs of arbitrary size and shape. The architecture we present generalizes standard molec…
Authors: David Duvenaud, Dougal Maclaurin, Jorge Aguilera-Iparraguirre
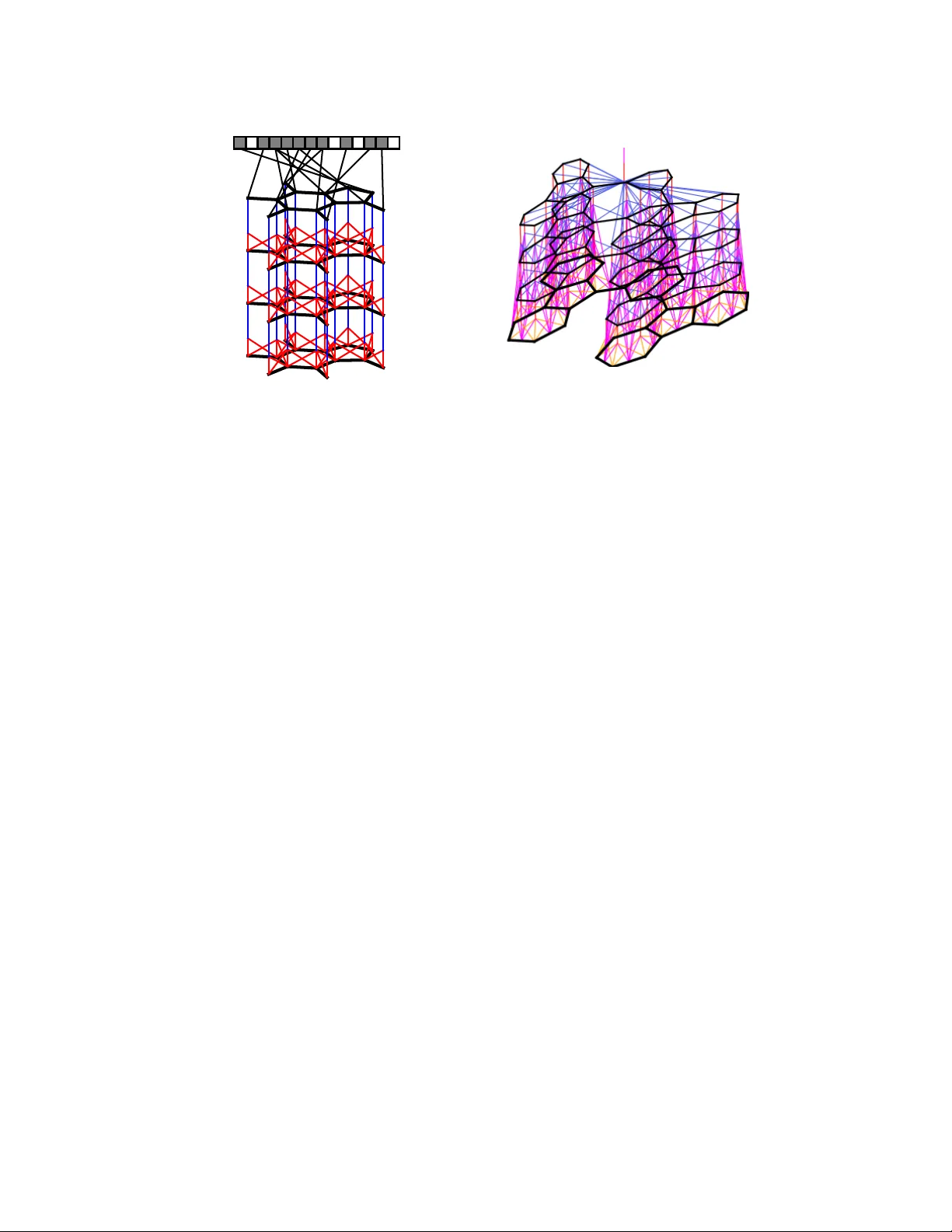
Con volutional Netw orks on Graphs f or Lear ning Molecular Fingerprints David Duv enaud † , Dougal Maclaurin † , Jorge Aguilera-Iparraguirr e Rafael G ´ omez-Bombarelli, T imothy Hirzel, Al ´ an Aspuru-Guzik, Ryan P . Adams Harvard Uni versity Abstract W e introduce a conv olutional neural network that operates directly on graphs. These networks allo w end-to-end learning of prediction pipelines whose inputs are graphs of arbitrary size and shape. The architecture we present generalizes standard molecular feature extraction methods based on circular fingerprints. W e show that these data-driven features are more interpretable, and have better pre- dictiv e performance on a v ariety of tasks. 1 Introduction Recent work in materials design used neural networks to predict the properties of nov el molecules by generalizing from examples. One dif ficulty with this task is that the input to the predictor , a molecule, can be of arbitrary size and shape. Currently , most machine learning pipelines can only handle inputs of a fixed size. The current state of the art is to use off-the-shelf fingerprint software to compute fixed-dimensional feature vectors, and use those features as inputs to a fully-connected deep neural network or other standard machine learning method. This formula was followed by [ 28 , 3 , 19 ]. During training, the molecular fingerprint vectors were treated as fixed. In this paper , we replace the bottom layer of this stack – the function that computes molecular fingerprint vectors – with a dif ferentiable neural network whose input is a graph representing the original molecule. In this graph, v ertices represent individual atoms and edges represent bonds. The lower layers of this network is con volutional in the sense that the same local filter is applied to each atom and its neighborhood. After several such layers, a global pooling step combines features from all the atoms in the molecule. These neural graph fingerprints offer se veral advantages o ver fix ed fingerprints: • Predicti ve performance. By using data adapting to the task at hand, machine-optimized fingerprints can pro vide substantially better predicti ve performance than fixed fingerprints. W e show that neural graph fingerprints match or beat the predicti ve performance of stan- dard fingerprints on solubility , drug efficac y , and organic photo voltaic ef ficiency datasets. • Parsimony . Fixed fingerprints must be extremely large to encode all possible substructures without ov erlap. For example, [ 28 ] used a fingerprint vector of size 43,000, after having remov ed rarely-occurring features. Dif ferentiable fingerprints can be optimized to encode only rele vant features, reducing downstream computation and regularization requirements. • Interpr etability . Standard fingerprints encode each possible fragment completely dis- tinctly , with no notion of similarity between fragments. In contrast, each feature of a neural graph fingerprint can be acti v ated by similar but distinct molecular fragments, making the feature representation more meaningful. † Equal contribution. 1 Figure 1: Left : A visual representation of the computational graph of both standard circular fin- gerprints and neural graph fingerprints. First, a graph is constructed matching the topology of the molecule being fingerprinted, in which nodes represent atoms, and edges represent bonds. At each layer , information flows between neighbors in the graph. Finally , each node in the graph turns on one bit in the fixed-length fingerprint vector . Right : A more detailed sketch including the bond information used in each operation. 2 Circular finger prints The state of the art in molecular fingerprints are extended-connectivity circular fingerprints (ECFP) [ 21 ]. Circular fingerprints [ 6 ] are a refinement of the Morgan algorithm [ 17 ], designed to encode which substructures are present in a molecule in a way that is in v ariant to atom-relabeling. Circular fingerprints generate each layer’ s features by applying a fix ed hash function to the concate- nated features of the neighborhood in the previous layer . The results of these hashes are then treated as integer indices, where a 1 is written to the fingerprint vector at the index given by the feature vector at each node in the graph. Figure 1 (left) sho ws a sketch of this computational architecture. Ignoring collisions, each index of the fingerprint denotes the presence of a particular substructure. The size of the substructures represented by each index depends on the depth of the network. Thus the number of layers is referred to as the ‘radius’ of the fingerprints. Circular fingerprints are analogous to con volutional networks in that they apply the same operation locally ev erywhere, and combine information in a global pooling step. 3 Creating a differ entiable fingerprint The space of possible network architectures is lar ge. In the spirit of starting from a known-good con- figuration, we designed a dif ferentiable generalization of circular fingerprints. This section describes our replacement of each discrete operation in circular fingerprints with a differentiable analog. Hashing The purpose of the hash functions applied at each layer of circular fingerprints is to combine information about each atom and its neighboring substructures. This ensures that any change in a fragment, no matter ho w small, will lead to a different fingerprint index being activ ated. W e replace the hash operation with a single layer of a neural network. Using a smooth function allows the acti vations to be similar when the local molecular structure v aries in unimportant ways. Indexing Circular fingerprints use an indexing operation to combine all the nodes’ feature vectors into a single fingerprint of the whole molecule. Each node sets a single bit of the fingerprint to one, at an index determined by the hash of its feature vector . This pooling-like operation con verts an arbitrary-sized graph into a fixed-sized vector . For small molecules and a lar ge fingerprint length, the fingerprints are alw ays sparse. W e use the softmax operation as a dif ferentiable analog of indexing. In essence, each atom is asked to classify itself as belonging to a single category . The sum of all these classification label vectors produces the final fingerprint. This operation is analogous to the pooling operation in standard con v olutional neural networks. 2 Algorithm 1 Circular fingerprints 1: Input: molecule, radius R , fingerprint length S 2: Initialize: fingerprint vector f ← 0 S 3: for each atom a in molecule 4: r a ← g ( a ) lookup atom features 5: for L = 1 to R for each layer 6: for each atom a in molecule 7: r 1 . . . r N = neighbors ( a ) 8: v ← [ r a , r 1 , . . . , r N ] concatenate 9: r a ← hash ( v ) hash function 10: i ← mod ( r a , S ) con vert to inde x 11: f i ← 1 Write 1 at index 12: Return: binary vector f Algorithm 2 Neural graph fingerprints 1: Input: molecule, radius R , hidden weights H 1 1 . . . H 5 R , output weights W 1 . . . W R 2: Initialize: fingerprint vector f ← 0 S 3: for each atom a in molecule 4: r a ← g ( a ) lookup atom features 5: for L = 1 to R for each layer 6: for each atom a in molecule 7: r 1 . . . r N = neighbors ( a ) 8: v ← r a + P N i =1 r i sum 9: r a ← σ ( v H N L ) smooth function 10: i ← softmax ( r a W L ) sparsify 11: f ← f + i add to fingerprint 12: Return: real-valued v ector f Figure 2: Pseudocode of circular fingerprints ( left ) and neural graph fingerprints ( right ). Differences are highlighted in blue. Every non-differentiable operation is replaced with a dif ferentiable analog. Canonicalization Circular fingerprints are identical regardless of the ordering of atoms in each neighborhood. This in variance is achie ved by sorting the neighboring atoms according to their features, and bond features. W e experimented with this sorting scheme, and also with applying the local feature transform on all possible permutations of the local neighborhood. An alternative to canonicalization is to apply a permutation-in variant function, such as summation. In the interests of simplicity and scalability , we chose summation. Circular fingerprints can be interpreted as a special case of neural graph fingerprints having lar ge random weights. This is because, in the limit of large input weights, tanh nonlinearities approach step functions, which when concatenated form a simple hash function. Also, in the limit of large input weights, the softmax operator approaches a one-hot-coded argmax operator , which is anal- ogous to an indexing operation. Algorithms 1 and 2 summarize these two algorithms and highlight their differences. Giv en a finger - print length L , and F features at each layer, the parameters of neural graph fingerprints consist of a separate output weight matrix of size F × L for each layer, as well as a set of hidden-to-hidden weight matrices of size F × F at each layer , one for each possible number of bonds an atom can hav e (up to 5 in organic molecules). 4 Experiments W e ran two experiments to demonstrate that neural fingerprints with large random weights behave similarly to circular fingerprints. First, we examined whether distances between circular fingerprints were similar to distances between neural fingerprint-based distances. Figure 3 (left) shows a scat- terplot of pairwise distances between circular vs. neural fingerprints. Fingerprints had length 2048, and were calculated on pairs of molecules from the solubility dataset [ 4 ]. Distance was measured using a continuous generalization of the T animoto (a.k.a. Jaccard) similarity measure, gi ven by distance ( x , y ) = 1 − X min( x i , y i ) . X max( x i , y i ) (1) There is a correlation of r = 0 . 823 between the distances. The line of points on the right of the plot shows that for some pairs of molecules, binary ECFP fingerprints ha ve exactly zero ov erlap. Second, we examined the predictive performance of neural fingerprints with large random weights vs. that of circular fingerprints. Figure 3 (right) shows av erage predictiv e performance on the sol- ubility dataset, using linear regression on top of fingerprints. The performances of both methods follow similar curv es. In contrast, the performance of neural fingerprints with small random weights follows a different curve, and is substantially better . This suggests that e ven with random weights, the relativ ely smooth acti vation of neural fingerprints helps generalization performance. 3 0.5 0.6 0.7 0.8 0.9 1.0 Circular fingerprint distances 0.5 0.6 0.7 0.8 0.9 1.0 Neural fingerprint distances N e u r a l v s C i r c u l a r d i s t a n c e s , r = 0 : 8 2 3 0 1 2 3 4 5 6 Fingerprint radius 0.8 1.0 1.2 1.4 1.6 1.8 2.0 RMSE (log Mol/L) Circular fingerprints Random conv with large parameters Random conv with small parameters Figure 3: Left: Comparison of pairwise distances between molecules, measured using circular fin- gerprints and neural graph fingerprints with large random weights. Right : Predictive performance of circular fingerprints (red), neural graph fingerprints with fixed large random weights (green) and neural graph fingerprints with fixed small random weights (blue). The performance of neural graph fingerprints with large random weights closely matches the performance of circular fingerprints. 4.1 Examining learned featur es T o demonstrate that neural graph fingerprints are interpretable, we sho w substructures which most activ ate individual features in a fingerprint vector . Each feature of a circular fingerprint vector can each only be activ ated by a single fragment of a single radius, except for accidental collisions. In contrast, neural graph fingerprint features can be activ ated by variations of the same structure, making them more interpretable, and allowing shorter feature v ectors. Solubility featur es Figure 4 sho ws the fragments that maximally acti v ate the most predicti ve fea- tures of a fingerprint. The fingerprint network was trained as inputs to a linear model predicting solubility , as measured in [ 4 ]. The feature sho wn in the top ro w has a positi ve predictive relationship with solubility , and is most acti vated by fragments containing a hydrophilic R-OH group, a standard indicator of solubility . The feature shown in the bottom row , strongly predictiv e of insolubility , is activ ated by non-polar repeated ring structures. Fragments most activ ated by pro-solubility feature O OH O NH O OH OH Fragments most activ ated by anti-solubility feature Figure 4: Examining fingerprints optimized for predicting solubility . Shown here are representati ve examples of molecular fragments (highlighted in blue) which most activ ate different features of the fingerprint. T op r ow: The feature most predictive of solubility . Bottom r ow: The feature most predictiv e of insolubility . 4 T oxicity features W e trained the same model architecture to predict toxicity , as measured in two different datasets in [ 26 ]. Figure 5 shows fragments which maximally activ ate the feature most predictiv e of toxicity , in two separate datasets. Fragments most activ ated by toxicity feature on SR-MMP dataset Fragments most activ ated by toxicity feature on NR-AHR dataset Figure 5: V isualizing fingerprints optimized for predicting toxicity . Shown here are representativ e samples of molecular fragments (highlighted in red) which most acti vate the feature most predicti ve of toxicity . T op r ow: the most predicti ve feature identifies groups containing a sulphur atom attached to an aromatic ring. Bottom r ow: the most predictiv e feature identifies fused aromatic rings, also known as polyc yclic aromatic hydrocarbons, a well-kno wn carcinogen. [ 27 ] constructed similar visualizations, but in a semi-manual way: to determine which toxic frag- ments activ ated a gi ven neuron, they searched ov er a hand-made list of toxic substructures and chose the one most correlated with a giv en neuron. In contrast, our visualizations are generated automati- cally , without the need to restrict the range of possible answers beforehand. 4.2 Predicti ve Perf ormance W e ran sev eral experiments to compare the predicti ve performance of neural graph fingerprints to that of the standard state-of-the-art setup: circular fingerprints fed into a fully-connected neural network. Experimental setup Our pipeline takes as input the SMILES [ 30 ] string encoding of each molecule, which is then con verted into a graph using RDKit [ 20 ]. W e also used RDKit to produce the extended circular fingerprints used in the baseline. Hydrogen atoms were treated implicitly . In our con volutional networks, the initial atom and bond features were chosen to be similar to those used by ECFP: Initial atom features concatenated a one-hot encoding of the atom’ s element, its degree, the number of attached hydrogen atoms, and the implicit valence, and an aromaticity indi- cator . The bond features were a concatenation of whether the bond type was single, double, triple, or aromatic, whether the bond was conjugated, and whether the bond was part of a ring. T raining and Architectur e T raining used batch normalization [ 11 ]. W e also experimented with tanh vs relu activ ation functions for both the neural fingerprint network layers and the fully- connected network layers. relu had a slight but consistent performance advantage on the valida- tion set. W e also experimented with dropconnect [ 29 ], a variant of dropout in which weights are randomly set to zero instead of hidden units, but found that it led to worse validation error in gen- eral. Each experiment optimized for 10000 minibatches of size 100 using the Adam algorithm [ 13 ], a variant of RMSprop that includes momentum. Hyperparameter Optimization T o optimize hyperparameters, we used random search. The hy- perparameters of all methods were optimized using 50 trials for each cross-v alidation fold. The following h yperparameters were optimized: log learning rate, log of the initial weight scale, the log L 2 penalty , fingerprint length, fingerprint depth (up to 6), and the size of the hidden layer in the fully-connected network. Additionally , the size of the hidden feature vector in the con volutional neural fingerprint networks was optimized. 5 Dataset Solubility [ 4 ] Drug efficac y [ 5 ] Photov oltaic ef ficiency [ 8 ] Units log Mol/L EC 50 in nM percent Predict mean 4.29 ± 0.40 1.47 ± 0.07 6.40 ± 0.09 Circular FPs + linear layer 1.71 ± 0.13 1.13 ± 0.03 2.63 ± 0.09 Circular FPs + neural net 1.40 ± 0.13 1.36 ± 0.10 2.00 ± 0.09 Neural FPs + linear layer 0.77 ± 0.11 1.15 ± 0.02 2.58 ± 0.18 Neural FPs + neural net 0.52 ± 0.07 1.16 ± 0.03 1.43 ± 0.09 T able 1: Mean predictiv e accuracy of neural fingerprints compared to standard circular fingerprints. Datasets W e compared the performance of standard circular fingerprints ag ainst neural graph fin- gerprints on a variety of domains: • Solubility: The aqueous solubility of 1144 molecules as measured by [ 4 ]. • Drug efficacy: The half-maximal effecti ve concentration (EC 50 ) in vitr o of 10,000 molecules against a sulfide-resistant strain of P . falciparum , the parasite that causes malaria, as measured by [ 5 ]. • Organic photov oltaic efficiency: The Harvard Clean Energy Project [ 8 ] uses expensiv e DFT simulations to estimate the photovoltaic efficienc y of organic molecules. W e used a subset of 20,000 molecules from this dataset. Predicti ve accuracy W e compared the performance of circular fingerprints and neural graph fin- gerprints under two conditions: In the first condition, predictions were made by a linear layer using the fingerprints as input. In the second condition, predictions were made by a one-hidden-layer neural network using the fingerprints as input. In all settings, all differentiable parameters in the composed models were optimized simultaneously . Results are summarized in T able 4.2 . In all experiments, the neural graph fingerprints matched or beat the accuracy of circular fingerprints, and the methods with a neural network on top of the fingerprints typically outperformed the linear layers. Software Automatic differentiation (AD) software packages such as Theano [ 1 ] significantly speed up dev elopment time by providing gradients automatically , b ut can only handle limited control structures and indexing. Since we required relativ ely complex control flo w and indexing in order to implement v ariants of Algorithm 2 , we used a more flexible automatic dif ferentiation package for Python called Autograd ( github.com/HIPS/autograd ). This package handles standard Numpy [ 18 ] code, and can dif ferentiate code containing while loops, branches, and indexing. Code for computing neural fingerprints and producing visualizations is av ailable at github.com/HIPS/neural-fingerprint . 5 Limitations Computational cost Neural fingerprints hav e the same asymptotic complexity in the number of atoms and the depth of the network as circular fingerprints, but hav e additional terms due to the matrix multiplies necessary to transform the feature vector at each step. T o be precise, computing the neural fingerprint of depth R , fingerprint length L of a molecule with N atoms using a molecular con v olutional net having F features at each layer costs O ( RN F L + RN F 2 ) . In practice, training neural networks on top of circular fingerprints usually took several minutes, while training both the fingerprints and the network on top took on the order of an hour on the lar ger datasets. Limited computation at each layer Ho w complicated should we make the function that goes from one layer of the network to the ne xt? In this paper we chose the simplest feasible architecture: a single layer of a neural network. Howe ver , it may be fruitful to apply multiple layers of nonlinear- ities between each message-passing step (as in [ 22 ]), or to make information preservation easier by adapting the Long Short-T erm Memory [ 10 ] architecture to pass information upwards. 6 Limited information pr opagation across the graph The local message-passing architecture de- veloped in this paper scales well in the size of the graph (due to the low degree of organic molecules), but its ability to propagate information across the graph is limited by the depth of the network. This may be appropriate for small graphs such as those representing the small organic molecules used in this paper . Ho wev er , in the w orst case, it can take a depth N 2 network to distinguish between graphs of size N . T o av oid this problem, [ 2 ] proposed a hierarchical clustering of graph substructures. A tree-structured network could examine the structure of the entire graph using only log ( N ) layers, but would require learning to parse molecules. T echniques from natural language processing [ 25 ] might be fruitfully adapted to this domain. Inability to distinguish stereoisomers Special bookkeeping is required to distinguish between stereoisomers, including enantomers (mirror images of molecules) and cis/trans isomers (rotation around double bonds). Most circular fingerprint implementations have the option to make these distinctions. Neural fingerprints could be e xtended to be sensiti ve to stereoisomers, but this remains a task for future work. 6 Related work This work is similar in spirit to the neural T uring machine [ 7 ], in the sense that we take an existing discrete computational architecture, and make each part differentiable in order to do gradient-based optimization. Neural nets f or quantitative structure-activity r elationship (QSAR) The modern standard for predicting properties of nov el molecules is to compose circular fingerprints with fully-connected neural networks or other re gression methods. [ 3 ] used circular fingerprints as inputs to an ensemble of neural networks, Gaussian processes, and random forests. [ 19 ] used circular fingerprints (of depth 2) as inputs to a multitask neural network, sho wing that multiple tasks helped performance. Neural graph fingerprints The most closely related work is [ 15 ], who build a neural network having graph-v alued inputs. Their approach is to remove all cycles and build the graph into a tree structure, choosing one atom to be the root. A recursi ve neural network [ 23 , 24 ] is then run from the leav es to the root to produce a fixed-size representation. Because a graph having N nodes has N possible roots, all N possible graphs are constructed. The final descriptor is a sum of the representations computed by all distinct graphs. There are as many distinct graphs as there are atoms in the network. The computational cost of this method thus grows as O ( F 2 N 2 ) , where F is the size of the feature vector and N is the number of atoms, making it less suitable for large molecules. Con volutional neural netw orks Con volutional neural networks ha ve been used to model images, speech, and time series [ 14 ]. Ho wev er , standard con volutional architectures use a fixed computa- tional graph, making them difficult to apply to objects of varying size or structure, such as molecules. More recently , [ 12 ] and others hav e de veloped a con volutional neural network architecture for mod- eling sentences of varying length. Neural networks on fixed graphs [ 2 ] introduce conv olutional networks on graphs in the regime where the graph structure is fix ed, and each training e xample differs only in ha ving dif ferent features at the vertices of the same graph. In contrast, our networks address the situation where each training input is a different graph. Neural networks on input-dependent graphs [ 22 ] propose a neural network model for graphs having an interesting training procedure. The forward pass consists of running a message-passing scheme to equilibrium, a fact which allo ws the re verse-mode gradient to be computed without storing the entire forward computation. They apply their network to predicting mutagenesis of molecular compounds as well as web page rankings. [ 16 ] also propose a neural network model for graphs with a learning scheme whose inner loop optimizes not the training loss, but rather the correlation between each newly-proposed vector and the training error residual. They apply their model to a dataset of boiling points of 150 molecular compounds. Our paper b uilds on these ideas, with the 7 following dif ferences: Our method replaces their complex training algorithms with simple gradient- based optimization, generalizes existing circular fingerprint computations, and applies these net- works in the context of modern QSAR pipelines which use neural networks on top of the fingerprints to increase model capacity . Unrolled inference algorithms [ 9 ] and others hav e noted that iterativ e inference procedures sometimes resemble the feedforward computation of a recurrent neural network. One natural exten- sion of these ideas is to parameterize each inference step, and train a neural network to approximately match the output of exact inference using only a small number of iterations. The neural fingerprint, when viewed in this light, resembles an unrolled message-passing algorithm on the original graph. 7 Conclusion W e generalized existing hand-crafted molecular features to allow their optimization for diverse tasks. By making each operation in the feature pipeline dif ferentiable, we can use standard neural-netw ork training methods to scalably optimize the parameters of these neural molecular fingerprints end-to- end. W e demonstrated the interpretability and predictiv e performance of these new fingerprints. Data-driv en features ha ve already replaced hand-crafted features in speech recognition, machine vision, and natural-language processing. Carrying out the same task for virtual screening, drug design, and materials design is a natural next step. Acknowledgments W e thank Edward Pyzer-Knapp, Jennifer W ei, and Samsung Adv anced Institute of T echnology for their support. This work was partially funded by NSF IIS-1421780. References [1] Fr ´ ed ´ eric Bastien, P ascal Lamblin, Razv an P ascanu, James Bergstra, Ian J. Goodfello w , Arnaud Bergeron, Nicolas Bouchard, and Y oshua Bengio. Theano: new features and speed improve- ments. Deep Learning and Unsupervised Feature Learning NIPS 2012 W orkshop, 2012. [2] Joan Bruna, W ojciech Zaremba, Arthur Szlam, and Y ann LeCun. Spectral networks and locally connected networks on graphs. arXiv pr eprint arXiv:1312.6203 , 2013. [3] George E. Dahl, Navdeep Jaitly , and Ruslan Salakhutdinov . Multi-task neural networks for QSAR predictions. arXiv pr eprint arXiv:1406.1231 , 2014. [4] John S. Delane y . ESOL: Estimating aqueous solubility directly from molecular structure. J our- nal of Chemical Information and Computer Sciences , 44(3):1000–1005, 2004. [5] Francisco-Javier Gamo, Laura M Sanz, Jaume V idal, Cristina de Cozar , Emilio Alvarez, Jose-Luis Lav andera, Dana E V anderwall, Darren VS Green, V inod Kumar , Samiul Hasan, et al. Thousands of chemical starting points for antimalarial lead identification. Natur e , 465(7296):305–310, 2010. [6] Robert C. Glem, Andreas Bender, Catrin H. Arnby , Lars Carlsson, Scott Boyer , and James Smith. Circular fingerprints: flexible molecular descriptors with applications from physical chemistry to ADME. IDrugs: the in vestigational drugs journal , 9(3):199–204, 2006. [7] Alex Graves, Greg W ayne, and Ivo Danihelka. Neural T uring machines. arXiv pr eprint arXiv:1410.5401 , 2014. [8] Johannes Hachmann, Roberto Oli vares-Amaya, Sule Atahan-Evrenk, Carlos Amador -Bedolla, Roel S S ´ anchez-Carrera, Aryeh Gold-Parker , Leslie V ogt, Anna M Brockway , and Al ´ an Aspuru-Guzik. The Harv ard clean ener gy project: large-scale computational screening and design of organic photo voltaics on the w orld community grid. The Journal of Physical Chem- istry Letters , 2(17):2241–2251, 2011. [9] John R Hershey , Jonathan Le Roux, and Felix W eninger . Deep unfolding: Model-based inspi- ration of nov el deep architectures. arXiv pr eprint arXiv:1409.2574 , 2014. 8 [10] Sepp Hochreiter and J ¨ urgen Schmidhuber . Long short-term memory . Neural computation , 9(8):1735–1780, 1997. [11] Serge y Ioffe and Christian Sze gedy . Batch normalization: Accelerating deep network training by reducing internal cov ariate shift. arXiv pr eprint arXiv:1502.03167 , 2015. [12] Nal Kalchbrenner, Edward Grefenstette, and Phil Blunsom. A con volutional neural network for modelling sentences. Pr oceedings of the 52nd Annual Meeting of the Association for Com- putational Linguistics , June 2014. [13] Diederik Kingma and Jimmy Ba. Adam: A method for stochastic optimization. arXiv pr eprint arXiv:1412.6980 , 2014. [14] Y ann LeCun and Y oshua Bengio. Conv olutional networks for images, speech, and time series. The handbook of brain theory and neural networks , 3361, 1995. [15] Alessandro Lusci, Gianluca Pollastri, and Pierre Baldi. Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules. Journal of chemical information and modeling , 53(7):1563–1575, 2013. [16] Alessio Micheli. Neural network for graphs: A contextual constructi ve approach. Neur al Networks, IEEE T ransactions on , 20(3):498–511, 2009. [17] H.L. Morg an. The generation of a unique machine description for chemical structure. Journal of Chemical Documentation , 5(2):107–113, 1965. [18] T ravis E Oliphant. Python for scientific computing. Computing in Science & Engineering , 9(3):10–20, 2007. [19] Bharath Ramsundar , Ste ven K earnes, Patrick Riley , Dale W ebster , Da vid K onerding, and V ijay Pande. Massively multitask netw orks for drug discovery . , 2015. [20] RDKit: Open-source cheminformatics. www.rdkit.org . [accessed 11-April-2013]. [21] David Rogers and Mathew Hahn. Extended-connectivity fingerprints. Journal of Chemical Information and Modeling , 50(5):742–754, 2010. [22] F . Scarselli, M. Gori, Ah Chung Tsoi, M. Hagenb uchner , and G. Monf ardini. The graph neural network model. Neural Networks, IEEE T ransactions on , 20(1):61–80, Jan 2009. [23] Richard Socher , Eric H Huang, Jeffrey Pennin, Christopher D Manning, and Andre w Y Ng. Dynamic pooling and unfolding recursi ve autoencoders for paraphrase detection. In Advances in Neural Information Processing Systems , pages 801–809, 2011. [24] Richard Socher , Jef frey Pennington, Eric H Huang, Andrew Y Ng, and Christopher D Man- ning. Semi-supervised recursive autoencoders for predicting sentiment distributions. In Pr o- ceedings of the Conference on Empirical Methods in Natural Language Pr ocessing , pages 151–161. Association for Computational Linguistics, 2011. [25] Kai Sheng T ai, Richard Socher , and Christopher D Manning. Impro ved semantic representations from tree-structured long short-term memory networks. arXiv pr eprint arXiv:1503.00075 , 2015. [26] T ox21 Challenge. National center for advancing translational sciences. http://tripod. nih.gov/tox21/challenge , 2014. [Online; accessed 2-June-2015]. [27] Thomas Unterthiner , Andreas Mayr , G ¨ unter Klambauer , and Sepp Hochreiter . T oxicity predic- tion using deep learning. arXiv pr eprint arXiv:1503.01445 , 2015. [28] Thomas Unterthiner , Andreas Mayr , G ¨ unter Klambauer , Marvin Steijaert, J ¨ org W enger , Hugo Ceulemans, and Sepp Hochreiter . Deep learning as an opportunity in virtual screening. In Advances in Neural Information Processing Systems , 2014. [29] Li W an, Matthew Zeiler , Sixin Zhang, Y ann L. Cun, and Rob Fer gus. Regularization of neural networks using dropconnect. In International Confer ence on Machine Learning , 2013. [30] David W eininger . SMILES, a chemical language and information system. Journal of chemical information and computer sciences , 28(1):31–36, 1988. 9
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment