Ab-Initio Study on the Hard Magnetic Properties of MnBi
We have studied the hard magnetic properties of the low-temperature phase of MnBi with first principle calculations based on the density functional theory. The calculations have been carried out on two distinct unit cell configurations MnBi and BiMn with the element in the unit cell origin named first. Our results show that these configurations are not equivalent and that MnBi describes the system better near T = 0K and the BiMn configuration describes the system better for T > 300K. The magnetic moments of both configurations agree well with experimental measurements considering both spin and orbital contributions. At high temperatures the magneto-crystalline anisotropy energy increases with increasing unit cell volume and reaches a maximum of 2:3MJ=m3 and a c=a ratio of 1:375.
💡 Research Summary
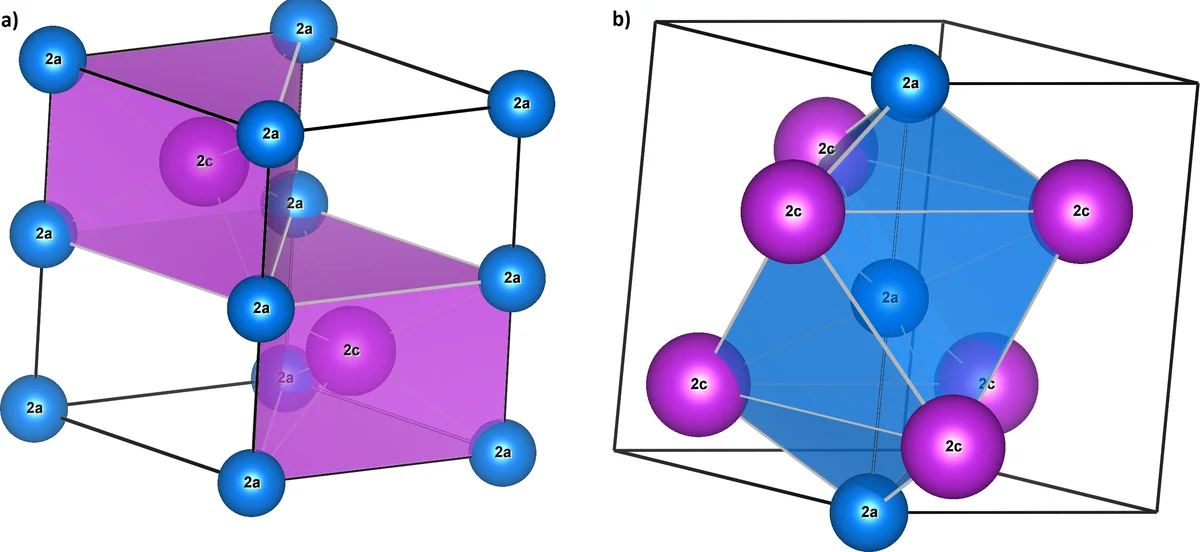
This paper presents a comprehensive first‑principles investigation of the low‑temperature phase (LTP) of MnBi, focusing on its unusual temperature‑dependent magnetic properties. The authors consider two distinct crystallographic configurations within the same space group (P6₃/mmc): “MnBi”, where Mn occupies the 2a Wyckoff position (the origin) and Bi occupies 2c, and “BiMn”, the inverse arrangement with Bi at 2a and Mn at 2c. Because the two sites have different local environments, swapping Mn and Bi creates two non‑equivalent electronic structures, which the authors hypothesize may dominate at different temperature regimes.
Density‑functional theory (DFT) calculations were carried out using the full‑potential augmented plane‑wave plus local orbitals (APW+lo) code WIEN2k, employing the Perdew‑Burke‑Ernzerhof generalized‑gradient approximation (PBE‑GGA) for exchange‑correlation. A dense k‑mesh of 25 × 25 × 15 and an energy convergence criterion of 10⁻⁹ Ry were used. After scalar‑relativistic spin‑polarized self‑consistency, spin‑orbit coupling (SOC) was introduced, and the magnetocrystalline anisotropy energy (MAE) was defined as MAE = E
Comments & Academic Discussion
Loading comments...
Leave a Comment