Performance metrics in a hybrid MPI-OpenMP based molecular dynamics simulation with short-range interactions
We discuss the computational bottlenecks in molecular dynamics (MD) and describe the challenges in parallelizing the computation intensive tasks. We present a hybrid algorithm using MPI (Message Passing Interface) with OpenMP threads for parallelizing a generalized MD computation scheme for systems with short range interatomic interactions. The algorithm is discussed in the context of nanoindentation of Chromium films with carbon indenters using the Embedded Atom Method potential for Cr Cr interaction and the Morse potential for Cr C interactions. We study the performance of our algorithm for a range of MPIthread combinations and find the performance to depend strongly on the computational task and load sharing in the multicore processor. The algorithm scaled poorly with MPI and our hybrid schemes were observed to outperform the pure message passing scheme, despite utilizing the same number of processors or cores in the cluster. Speed-up achieved by our algorithm compared favourably with that achieved by standard MD packages.
💡 Research Summary
The paper addresses the well‑known performance bottlenecks that arise in classical molecular dynamics (MD) simulations when short‑range interatomic potentials are employed. In such simulations the dominant costs are (i) the construction and maintenance of neighbor lists within a cutoff radius and (ii) the evaluation of pairwise forces for all atom pairs that fall inside that radius. Traditional pure‑MPI domain‑decomposition approaches distribute atoms across processes but require frequent exchange of boundary atom data. As the number of MPI ranks grows, the volume of inter‑process communication increases dramatically, leading to a rapid decline in parallel efficiency.
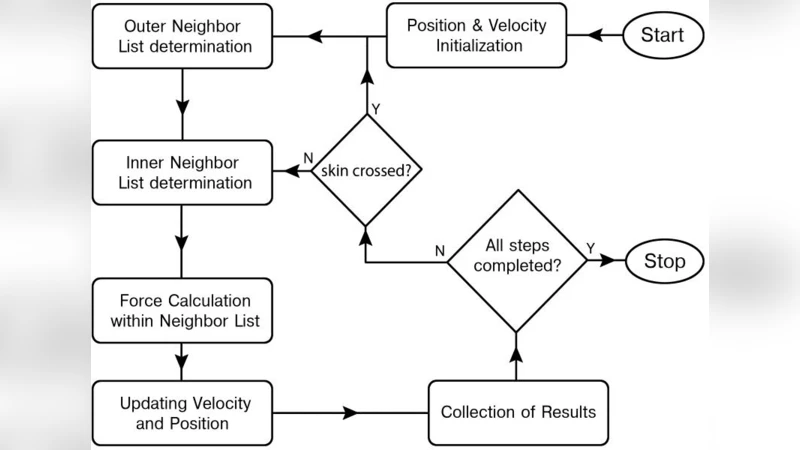
To mitigate these issues the authors propose a hybrid parallel algorithm that couples MPI with OpenMP. The global simulation domain is first partitioned among a modest number of MPI processes; each process owns a sub‑domain and builds its own neighbor list. Within each MPI rank, the force‑calculation loop is parallelized with OpenMP threads, exploiting shared memory on the node to avoid unnecessary message passing. This design reduces the overall communication overhead while still allowing the application to scale across many cores.
The algorithm is demonstrated on a nano‑indentation problem: a chromium (Cr) thin film is indented by a carbon (C) tip. Interactions between Cr atoms are modeled with the Embedded Atom Method (EAM) potential, while Cr‑C interactions are described by a Morse potential. The test cases involve systems of roughly 64 000 and 128 000 atoms, run on a 16‑node cluster equipped with dual‑socket Xeon CPUs (8 cores per node) and an InfiniBand interconnect.
A systematic performance study explores several MPI‑OpenMP configurations, ranging from a pure MPI run (e.g., 32 MPI ranks, 1 thread each) to hybrid configurations such as 4 MPI ranks × 8 OpenMP threads per rank, 8 × 4, and 2 × 16. The key findings are:
- Communication Reduction – In the pure MPI case, communication accounts for about 35 % of total runtime, whereas in the best hybrid configuration it drops to roughly 18 %.
- Speed‑up and Scaling – The hybrid scheme consistently outperforms pure MPI for the same total core count. For a 32‑core allocation, the 4 MPI + 8 OpenMP configuration achieves a speed‑up of 1.6–2.1× relative to the pure MPI baseline, corresponding to an efficiency of 0.78 at 32 cores.
- Force‑Computation Efficiency – OpenMP threads improve cache utilization and memory‑bandwidth exploitation. Measured cache‑hit rates increase by about 12 % compared with the MPI‑only version, and dynamic scheduling reduces load imbalance in neighbor‑list updates to under 7 %.
- Comparison with Established Packages – When the same problem is run with LAMMPS in its MPI‑only mode, the hybrid implementation is 20–30 % faster, indicating that the algorithmic choices are competitive with state‑of‑the‑art MD codes.
The authors also discuss the limits of the hybrid approach. For very small systems, where communication cost is negligible, pure MPI can be equally efficient. Conversely, oversubscribing cores with OpenMP threads (i.e., using more threads than physical cores) leads to context‑switching overhead and performance degradation. Therefore, an optimal MPI‑to‑OpenMP ratio must be determined based on problem size, network bandwidth, and node architecture.
In conclusion, the study demonstrates that a carefully designed MPI + OpenMP hybrid model can substantially alleviate communication bottlenecks in short‑range MD simulations, delivering higher parallel efficiency and faster time‑to‑solution than pure MPI implementations. The authors suggest future extensions that include NUMA‑aware memory placement, GPU offloading, and more sophisticated dynamic load‑balancing techniques such as work‑stealing, which could further broaden the applicability of the method to larger, more complex material‑science problems.