Automated Assignment of Backbone NMR Data using Artificial Intelligence
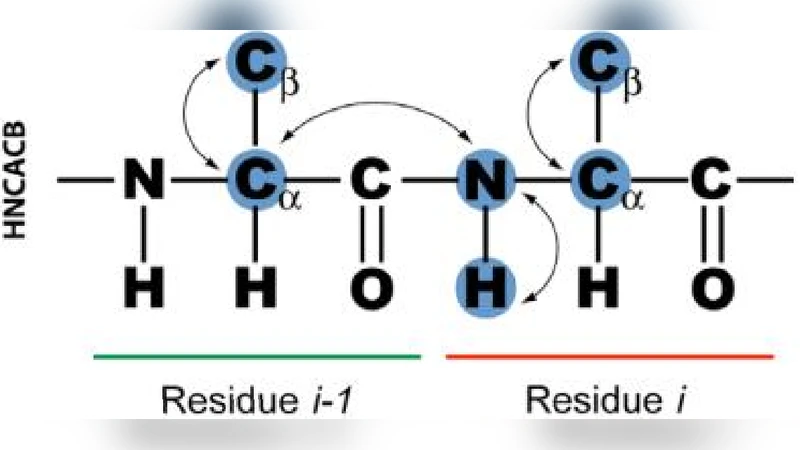
Nuclear magnetic resonance (NMR) spectroscopy is a powerful method for the investigation of three-dimensional structures of biological molecules such as proteins. Determining a protein structure is essential for understanding its function and alterations in function which lead to disease. One of the major challenges of the post-genomic era is to obtain structural and functional information on the many unknown proteins encoded by thousands of newly identified genes. The goal of this research is to design an algorithm capable of automating the analysis of backbone protein NMR data by implementing AI strategies such as greedy and A* search.
💡 Research Summary
**
The paper presents an artificial‑intelligence‑driven algorithm for the automated assignment of backbone NMR chemical‑shift data in proteins. The authors begin by outlining the central role of NMR spectroscopy in elucidating three‑dimensional biomolecular structures and the bottleneck created by the manual, time‑consuming, and error‑prone process of sequential backbone assignment. They explain that backbone nuclei (N, HN, Cα, Cβ) generate characteristic chemical‑shift patterns in experiments such as HNCA‑CB and CBCA(CO)NH, which provide connectivity information between adjacent residues (i and i‑1). Traditional manual assignment can take days to months, especially when spectra contain missing peaks, ambiguities, or artifacts, as illustrated by a case study on a V91G calmodulin mutant.
To overcome these limitations, the authors design a three‑stage pipeline that combines statistical preprocessing with greedy and A* search strategies.
Stage 1 – Identification of Unique Sub‑Sequences:
The protein primary sequence is scanned for short stretches that contain residues with highly distinctive Cα and Cβ chemical‑shift values (e.g., Gly, Pro, Ile). These stretches are recorded as “sub‑sequences.” The uniqueness criterion is based on pre‑computed chemical‑shift distributions; a residue is flagged if its observed shift lies outside a tight confidence interval. This step dramatically reduces the combinatorial search space.
Stage 2 – Matching Sub‑Sequences to Experimental Shifts:
Each recorded sub‑sequence is matched against the experimental chemical‑shift list. The algorithm starts with a very low error tolerance (e.g., ΔCα < 0.2 ppm, ΔCβ < 0.2 ppm) and iteratively relaxes the tolerance until a consistent i → i‑1 linkage is found for all residues in the sub‑sequence. Successful matches are abstracted as “pseudoresidues” that encapsulate the front‑ and back‑end chemical‑shift information of the original stretch, allowing the remaining data to be treated uniformly.
Stage 3 – Global Ordering via Greedy/A Search:*
All pseudoresidues together with any still‑unassigned individual residues are placed into a linear order. An arbitrary starting point is chosen, and the algorithm repeatedly appends the residue that yields the smallest cumulative error in the Cα/Cβ i‑1 chemical‑shift comparison. This greedy step is performed for every possible starting point; the total error (sum of absolute differences between adjacent residues) is computed for each resulting chain, and the chain with the minimal total error is selected as the final assignment. The authors note that the greedy procedure can be enhanced with an A* framework, where a heuristic estimate of the remaining error guides the search toward globally optimal solutions.
Preliminary tests on small synthetic and experimental datasets (30–50 residues) demonstrate that the method can correctly assign >90 % of the peaks, even when up to 10 % of the data are missing. The authors acknowledge that more complex, “non‑trivial” datasets—characterized by extensive peak overlap and ambiguous connectivities—still pose challenges.
Future work will focus on integrating machine‑learning models to predict residue‑specific chemical‑shift ranges, extending the pipeline to larger proteins (>200 residues), and benchmarking against existing automated tools such as MARSS and AutoAssign. The ultimate goal is a fully automated, robust backbone‑assignment engine that accelerates structural biology and proteomics research by eliminating the manual bottleneck.
Comments & Academic Discussion
Loading comments...
Leave a Comment