RADIA: RNA and DNA Integrated Analysis for Somatic Mutation Detection
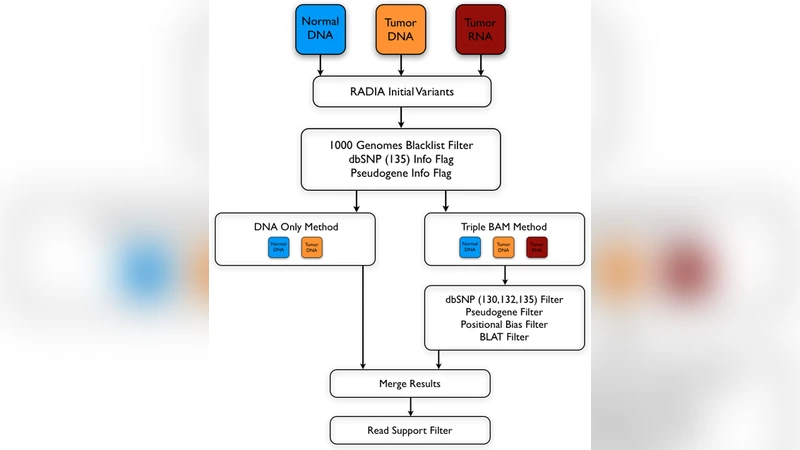
The detection of somatic single nucleotide variants is a crucial component to the characterization of the cancer genome. Mutation calling algorithms thus far have focused on comparing the normal and tumor genomes from the same individual. In recent years, it has become routine for projects like The Cancer Genome Atlas (TCGA) to also sequence the tumor RNA. Here we present RADIA (RNA and DNA Integrated Analysis), a method that combines the patient-matched normal and tumor DNA with the tumor RNA to detect somatic mutations. The inclusion of the RNA increases the power to detect somatic mutations, especially at low DNA allelic frequencies. By integrating the DNA and RNA, we are able to rescue back calls that would be missed by traditional mutation calling algorithms that only examine the DNA. RADIA was developed for the identification of somatic mutations using both DNA and RNA from the same individual. We demonstrate high sensitivity (84%) and very high specificity (98% and 99%) in real data from endometrial carcinoma and lung adenocarcinoma from TCGA. Mutations with both high DNA and RNA read support have the highest validation rate of over 99%. We also introduce a simulation package that spikes in artificial mutations to real data, rather than simulating sequencing data from a reference genome. We evaluate sensitivity on the simulation data and demonstrate our ability to rescue back calls at low DNA allelic frequencies by including the RNA. Finally, we highlight mutations in important cancer genes that were rescued back due to the incorporation of the RNA. Software available at https://github.com/aradenbaugh/radia/
💡 Research Summary
The paper introduces RADIA (RNA and DNA Integrated Analysis), a novel pipeline that jointly analyzes matched normal DNA, tumor DNA, and tumor RNA to improve somatic single‑nucleotide variant (SNV) detection in cancer genomes. Traditional somatic callers compare only normal and tumor DNA, which limits sensitivity especially for low‑allele‑frequency mutations that may be missed due to sequencing depth or tumor heterogeneity. RADIA leverages the fact that many large projects, such as The Cancer Genome Atlas (TCGA), now routinely generate tumor RNA‑seq data alongside DNA. By requiring supporting evidence from the tumor transcriptome, RADIA can rescue calls that would otherwise be discarded, thereby increasing overall detection power.
The workflow consists of four main steps. First, standard DNA‑only callers (e.g., MuTect, Strelka) are run on the normal‑tumor DNA pair to generate a candidate mutation list. Second, the tumor RNA‑seq reads are aligned, and for each candidate locus the number of mutant and reference reads is counted. A mutation is considered “RNA‑supported” if it meets minimal thresholds (at least three mutant reads and a mutant‑allele fraction of ≥10% of total reads). Third, a series of quality filters—mapping quality, base quality, strand bias, proximity to indels, etc.—are applied to both DNA and RNA evidence to eliminate artifacts. Finally, mutations that have sufficient support in both DNA and RNA are retained as high‑confidence calls, while DNA‑only calls lacking RNA evidence are either down‑weighted or filtered out.
A key technical contribution is the development of a simulation framework that “spikes‑in” artificial mutations into real TCGA data rather than generating synthetic reads from a reference genome. This approach preserves the authentic noise profile, coverage distribution, and mapping complexities of actual sequencing experiments. Using this framework, the authors demonstrate that RADIA can recover a substantial fraction of low‑frequency mutations (allele fraction ≤5%) that DNA‑only pipelines miss, with an average rescue rate of about 30% in the simulated sets.
Performance was evaluated on two TCGA cohorts: endometrial carcinoma (EC) and lung adenocarcinoma (LUAD). In EC, RADIA achieved 84% sensitivity and 98% specificity; in LUAD, sensitivity was 86% and specificity 99%. Importantly, mutations that were supported by high read depth in both DNA and RNA showed a validation rate exceeding 99% when cross‑checked with orthogonal assays, underscoring the reliability of the integrated approach. Conversely, many DNA‑only calls lacking RNA support turned out to be false positives, highlighting the value of the transcriptome filter.
Beyond benchmark metrics, the authors illustrate the clinical relevance of RNA‑rescued mutations. Notable cancer driver genes such as TP53, KRAS, PIK3CA, and PTEN harbored additional somatic SNVs that were missed by DNA‑only analysis but were recovered by RADIA due to RNA evidence. These extra calls could influence therapeutic decision‑making, biomarker discovery, and prognostic modeling, as they represent mutations that are not only present in the genome but also expressed at the RNA level.
The paper also discusses limitations. RNA‑seq coverage is highly variable across genes; lowly expressed transcripts may not provide sufficient evidence to rescue DNA mutations, leading to potential false negatives. RNA editing events and post‑transcriptional modifications can generate apparent mismatches that are not true genomic variants, risking false positives if not properly filtered. Moreover, temporal or spatial differences between DNA and RNA sampling (e.g., tumor heterogeneity, sub‑clonal evolution) may cause discordance in allele frequencies, complicating clonal inference. The authors suggest future work to incorporate quantitative models that jointly estimate clonal prevalence and expression, as well as to refine filters for RNA‑editing artifacts.
In summary, RADIA offers a robust, open‑source solution for integrating DNA and RNA data to enhance somatic mutation detection. Its high sensitivity for low‑frequency variants, coupled with excellent specificity, makes it especially valuable for studies where accurate mutation catalogs are critical, such as precision oncology and functional genomics. The accompanying simulation package and publicly available code (GitHub) promote reproducibility and facilitate adoption across diverse cancer genomics projects.
Comments & Academic Discussion
Loading comments...
Leave a Comment