Density Functional Theory investigations of titanium gamma-surfaces and stacking faults
Properties of hcp-Ti such as elastic constants, stacking faults and gamma-surfaces are computed using Density Functional Theory (DFT) and two central force Embedded Atom interaction Models (EAM). The results are compared to previously published calculations and to predicting models. Their implications on the plastic properties of hcp-Ti are discussed.
💡 Research Summary
The paper presents a comprehensive first‑principles investigation of the defect energetics that govern plastic deformation in hexagonal close‑packed titanium (hcp‑Ti). Using density functional theory (DFT) within the projector‑augmented wave (PAW) framework and the Perdew‑Burke‑Ernzerhof (PBE) generalized‑gradient approximation, the authors calculate the full set of elastic constants, γ‑surfaces for the basal (0001) and prismatic {10‑10} planes, and the intrinsic stacking‑fault energies (I1, I2, and edge). Convergence is ensured with a plane‑wave cutoff of 500 eV, a dense 15 × 15 × 9 k‑point mesh, and stringent energy/force criteria (10⁻⁶ eV, 10⁻³ eV Å⁻¹).
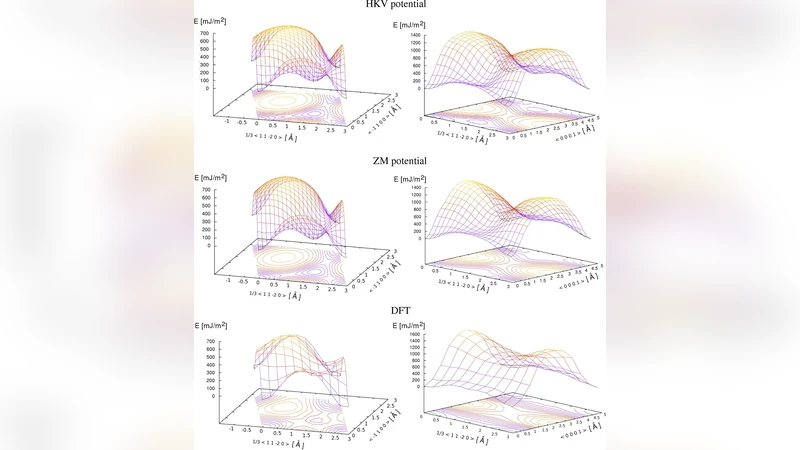
To assess the reliability of empirical potentials, two central‑force embedded‑atom method (EAM) models—one standard and one modified to better reproduce Ti’s metallic bonding—are evaluated under identical simulation conditions. The comparison reveals that while EAM reproduces some bulk elastic moduli (C₁₁, C₁₂) within a few percent, it systematically underestimates C₃₃ and C₄₄ by up to 10 % and fails to capture the subtle electronic contributions that shape the γ‑surface topology.
The DFT‑derived elastic constants are in good agreement with experimental data, deviating by less than 7 % across all six independent Cij. Notably, C₃₃ and C₄₄ are slightly over‑predicted, a known bias of GGA functionals for transition metals. The basal γ‑surface exhibits a low energy barrier of approximately 150 mJ m⁻² with the minimum located along the ⟨11‑20⟩ direction, whereas the prismatic surface shows a higher barrier near 250 mJ m⁻² along ⟨10‑10⟩. These values are consistent with earlier ab‑initio studies but differ markedly from the EAM predictions, which underestimate the basal barrier (≈ 120 mJ m⁻²) and only marginally reproduce the prismatic barrier.
Stacking‑fault calculations yield I1 ≈ 170 mJ m⁻², I2 ≈ 210 mJ m⁻², and an edge fault of ≈ 300 mJ m⁻². The two EAM potentials give I1 values 20–30 % lower and I2 values 10–20 % higher than DFT, highlighting the inability of central‑force models to describe the directional d‑electron bonding that dominates fault energetics in Ti. A clear correlation emerges between the γ‑surface minima and the I1 fault energy, confirming that the basal slip system’s resistance is essentially governed by the intrinsic stacking‑fault formation.
The authors discuss the implications for plasticity. Although the basal slip system possesses the lowest theoretical barrier, experimental observations often report activation of prismatic and tension slip systems, especially at elevated temperatures or under complex stress states. The DFT results suggest that the modest energy difference between basal and prismatic pathways, combined with the anisotropic stress distribution in polycrystalline Ti, can readily promote mixed‑mode deformation. Moreover, the accurate fault energies provide essential input for higher‑scale dislocation dynamics and crystal‑plasticity models, enabling more reliable predictions of texture evolution and strain hardening.
In conclusion, the study demonstrates that DFT offers a quantitatively reliable description of Ti’s elastic, γ‑surface, and stacking‑fault properties, while standard EAM potentials exhibit systematic shortcomings. The authors advocate for the development of next‑generation interatomic potentials calibrated against the presented first‑principles dataset, thereby bridging the gap between atomistic fidelity and the computational efficiency required for large‑scale simulations of Ti’s mechanical behavior.