Simulations of HIV capsid protein dimerization reveal the effect of chemistry and topography on the mechanism of hydrophobic protein association
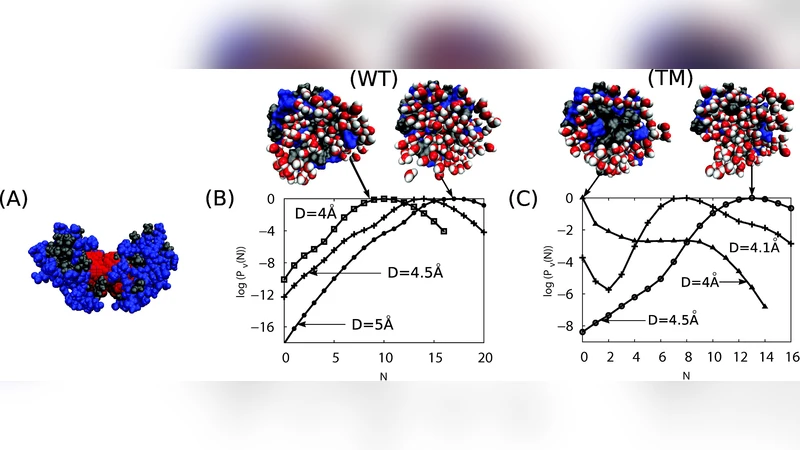
Recent work has shown that the hydrophobic protein surfaces in aqueous solution sit near a drying transition. The tendency for these surfaces to expel water from their vicinity leads to self assembly of macromolecular complexes. In this article we show with a realistic model for a biologically pertinent system how this phenomenon appears at the molecular level. We focus on the association of the C-terminal domain (CA-C) of the human immunodeficiency virus (HIV) capsid protein. By combining all-atom simulations with specialized sampling techniques we measure the water density distribution during the approach of two CA-C proteins as a function of separation and amino acid sequence in the interfacial region. The simulations demonstrate that CA-C protein-protein interactions sit at the edge of a dewetting transition and that this mesoscopic manifestation of the underlying liquid-vapor phase transition can be readily manipulated by biology or protein engineering to significantly affect association behavior. While the wild type protein remains wet until contact, we identify a set of in silico mutations, in which three hydrophilic amino acids are replaced with nonpolar residues, that leads to dewetting prior to association. The existence of dewetting depends on the size and relative locations of substituted residues separated by nm length scales, indicating long range cooperativity and a sensitivity to surface topography. These observations identify important details which are missing from descriptions of protein association based on buried hydrophobic surface area.
💡 Research Summary
The paper investigates how water-mediated dewetting influences the association of the C‑terminal domain of the HIV capsid protein (CA‑C) at an atomic level. Building on the hypothesis that hydrophobic protein surfaces in aqueous solution sit near a drying transition, the authors combine all‑atom molecular dynamics with specialized sampling techniques—specifically indirect umbrella sampling (INDUS) and umbrella‑type potentials—to map the water density distribution as two CA‑C monomers approach each other. By varying the inter‑monomer separation and systematically mutating residues in the interfacial region, they quantify how the local water environment changes during the early stages of dimerization.
For the wild‑type protein, simulations show that water remains trapped in the interface until the monomers are essentially in contact (≈0.6 nm separation). The presence of several hydrophilic residues (e.g., Asp61, Glu62, Lys63) forms strong hydrogen bonds with water, preventing premature drying. In contrast, a set of in‑silico mutations that replace these three hydrophilic side chains with non‑polar residues (valine, leucine, phenylalanine, etc.) triggers dewetting already at ≈0.8 nm separation. The dewetting transition is abrupt, indicating that the interface has crossed a mesoscopic analogue of a liquid‑vapor phase transition.
A key insight is that dewetting does not depend solely on the total non‑polar surface area; it is highly sensitive to the size, shape, and spatial arrangement of the mutated residues. When the same volume is occupied by small non‑polar groups such as alanine, no dewetting occurs, whereas larger groups (e.g., phenylalanine or tryptophan) placed at strategic positions promote water expulsion. This demonstrates a long‑range cooperativity: residues separated by nanometer‑scale distances can collectively lower the free‑energy barrier for drying, effectively creating a “drying nucleus” that spreads across the interface.
To assess the thermodynamic impact, the authors compute potentials of mean force (PMFs) for both wild‑type and mutant dimers. The mutant exhibits a reduction of the binding free‑energy barrier by roughly 2–3 kcal mol⁻¹ relative to the wild type, implying a substantially faster association rate. The analysis thus links the microscopic water density fluctuations directly to macroscopic binding kinetics.
Beyond the specific system, the study challenges the conventional view that buried hydrophobic surface area alone predicts protein‑protein affinity. It shows that water’s collective behavior, modulated by subtle changes in surface topography and chemistry, can dominate the early association pathway. The findings suggest new design principles for protein engineering: by judiciously altering side‑chain size and placement, one can tune the propensity for dewetting and thereby control assembly rates and stability. This has implications for antiviral strategies targeting capsid assembly, for the design of synthetic nanocontainers, and for any application where precise control of protein‑protein interfaces is required.
In summary, the work provides compelling computational evidence that CA‑C dimerization sits at the edge of a dewetting transition, that this transition can be switched on or off by a handful of targeted mutations, and that the effect propagates over nanometer distances due to cooperative surface topography. It bridges the gap between molecular‑scale water physics and biologically relevant protein association, offering a richer framework for understanding and engineering macromolecular assembly.
Comments & Academic Discussion
Loading comments...
Leave a Comment