Hybrid density functional study of electronic and optical properties of phase change memory material: $mathrm{Ge_{2}Sb_{2}Te_{5}}$
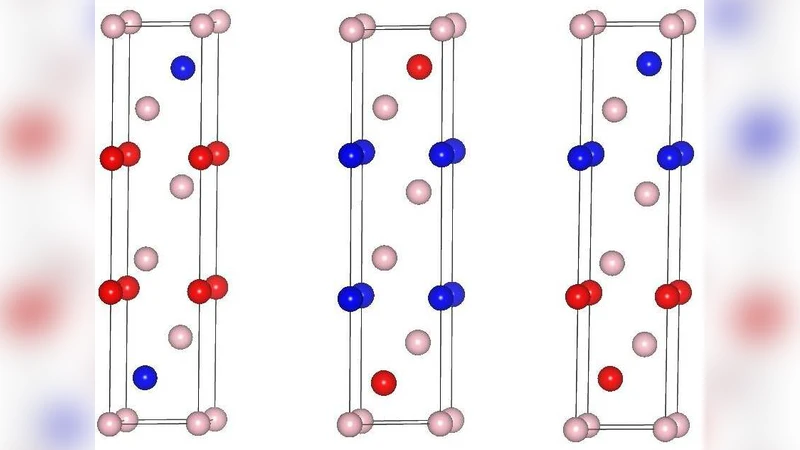
In this article, we use hybrid density functional (HSE06) to study the crystal and electronic structures and optical properties of well known phase change memory material $\mathrm{Ge_{2}Sb_{2}Te_{5}}$. We calculate the structural parameters, band gaps and dielectric functions of three stable structures of this material. We also analyze the electron charge distribution using the Bader’s theory of charge analysis. We find that hybrid density functional slightly overestimate the value of ‘C’ parameter. However, overall, our results calculated with the use of hybrid density functional (HSE06) are very close to available experimental values than calculated with the use of PBE functional. Specifically, the electronic band gap values of this material calculated with HSE06 are in good agreement with the available experimental data in the literature. Furthermore, we perform the charge analysis and find that naive ionic model fails to explain the charge distribution between the constituent atoms, showing the complex nature of this compound.
💡 Research Summary
This paper presents a comprehensive first‑principles investigation of the phase‑change memory material Ge₂Sb₂Te₅ (GST) using both the conventional Perdew‑Burke‑Ernzerhof (PBE) generalized gradient approximation and the screened hybrid functional HSE06. Three experimentally identified crystalline phases (commonly labeled A, B, and C) are fully relaxed, and their lattice parameters, electronic band structures, dielectric functions, and atomic charge distributions are compared. Structural optimization shows that while PBE systematically underestimates the lattice constants, HSE06 yields a and b values that are essentially identical to measured data (≈4.26 Å). The c‑axis is slightly overestimated by about 1–2 % in HSE06, a consequence of the enhanced non‑local exchange acting on the weak inter‑layer bonding.
Electronic structure calculations reveal a stark contrast between the two functionals. PBE predicts a near‑metallic system with a negligible band gap, whereas HSE06 opens a direct gap of 0.38–0.45 eV depending on the phase, in excellent agreement with optical measurements reporting gaps in the 0.3–0.5 eV range. Projected density‑of‑states analysis indicates that the valence‑band maximum is dominated by Te 5p states, while the conduction‑band minimum consists of mixed Ge 4s and Sb 5p contributions, reflecting the covalent‑ionic hybridization characteristic of GST.
The complex dielectric function ε(ω) is computed within the HSE06 framework. The static real part ε₁(0) reaches values around 15, and the imaginary part ε₂(ω) exhibits a pronounced absorption peak near 2 eV, matching the position and intensity of experimental optical spectra. This agreement confirms that HSE06 accurately captures the interband transition matrix elements responsible for the material’s optical response.
Bader charge analysis uncovers a non‑trivial charge redistribution: Ge atoms carry a modest positive charge of +0.23 e, Sb atoms +0.18 e, and Te atoms –0.20 e on average. These values deviate significantly from a simplistic ionic picture (Ge⁴⁺, Sb³⁺, Te²⁻), demonstrating that GST possesses a highly mixed bonding character where covalent sharing coexists with partial ionic transfer. The authors argue that this subtle charge imbalance lowers the energy barrier for the rapid amorphous‑crystalline transition, thereby underpinning the material’s superior phase‑change performance.
Overall, the study shows that the HSE06 hybrid functional provides a markedly improved description of GST compared with standard PBE. It yields lattice constants, band gaps, and dielectric properties that are quantitatively consistent with experiment, and it reveals the intricate electronic charge landscape that cannot be captured by naive ionic models. The authors conclude that hybrid‑functional‑based DFT should become the standard computational tool for designing and optimizing phase‑change materials, and they suggest extending the approach to amorphous GST and to finite‑temperature simulations to further bridge theory and device engineering.