Molecular dynamics beyonds the limits: massive scaling on 72 racks of a BlueGene/P and supercooled glass transition of a 1 billion particles system
We report scaling results on the world’s largest supercomputer of our recently developed Billions-Body Molecular Dynamics (BBMD) package, which was especially designed for massively parallel simulations of the atomic dynamics in structural glasses and amorphous materials. The code was able to scale up to 72 racks of an IBM BlueGene/P, with a measured 89% efficiency for a system with 100 billion particles. The code speed, with less than 0.14 seconds per iteration in the case of 1 billion particles, paves the way to the study of billion-body structural glasses with a resolution increase of two orders of magnitude with respect to the largest simulation ever reported. We demonstrate the effectiveness of our code by studying the liquid-glass transition of an exceptionally large system made by a binary mixture of 1 billion particles.
💡 Research Summary
This paper presents the development, optimization, and large‑scale deployment of the Billions‑Body Molecular Dynamics (BBMD) code, specifically engineered for simulating the atomic dynamics of structural glasses and amorphous materials at unprecedented system sizes. The authors target the IBM BlueGene/P architecture, scaling the application up to the full machine at the Jülich Supercomputing Centre—72 racks comprising 294,912 cores (≈1 PFLOP peak performance).
Algorithmic design: BBMD implements short‑range Lennard‑Jones and soft‑sphere potentials, truncated smoothly at a cutoff distance r_c to limit computational effort. The simulation domain is decomposed into cubic cells of size r_c, and each cell maintains a bidirectional linked list containing particle attributes (position, velocity, acceleration, mass, species). This structure enables O(N) neighbor searching without the memory‑intensive neighbor‑list approach used in many traditional MD codes. By moving particles between cells via pointer manipulation rather than allocation/deallocation, memory bandwidth is conserved and cache utilization is maximized.
Parallelization strategy: The code adopts a spatial domain decomposition (one MPI rank per sub‑domain) and relies on nearest‑neighbor communications only. Although a naïve implementation would require 26 messages per rank (the 26 surrounding cells in 3‑D), BBMD reduces the communication count to six by enlarging the communication window and exchanging ghost‑cell data in bulk. Particle migration across domain boundaries is handled by tagging particles (flipping the sign of their mass) and sorting them on‑the‑fly, which groups outbound particles contiguously for efficient MPI_Send/Recv. The communication phase is launched before particle exchange, allowing the subsequent force‑calculation phase to overlap with data transfer, thereby minimizing MPI wait times.
BlueGene‑specific optimizations: The most expensive operation in force evaluation is the square‑root required for distance computation. BBMD replaces the generic sqrt call with the BlueGene hardware reciprocal‑sqrt function (frsqrte) followed by two Newton‑Raphson iterations, achieving roughly a 7 % speedup. Additional low‑level tuning (alignment of data structures, avoidance of unnecessary branching) further improves throughput.
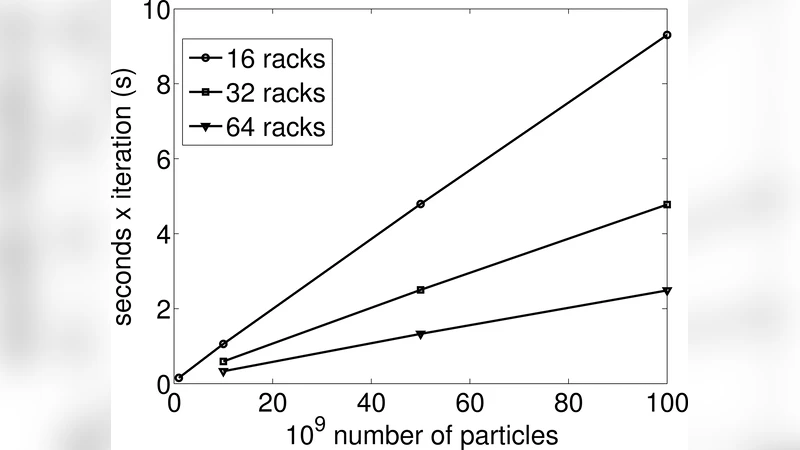
Scaling results – strong scaling: Benchmarks were performed for three system sizes: 1 × 10⁹, 1 × 10¹⁰, and 1 × 10¹¹ particles. For the 1 billion‑particle case, the code achieved 68 % parallel efficiency on 16 racks relative to a single rack, with a per‑step wall‑clock time of 0.14 s. Scaling to 10 billion particles yielded 70 % efficiency on 64 racks, while the 100 billion‑particle run reached 89 % efficiency on the full 72‑rack machine.
Weak scaling: Keeping the core count fixed while increasing particle number from 1 billion to 100 billion demonstrated near‑perfect linear O(N) scaling. The ratio of computation to communication time grew with problem size; for the largest runs, communication accounted for less than 1 % of total runtime, confirming that the three‑dimensional domain decomposition and overlapped communication strategy effectively eliminate communication bottlenecks at extreme scale.
Scientific application – glass transition: To validate the scientific utility of BBMD, the authors simulated a binary mixture of soft spheres (80 % type‑1, 20 % type‑2) comprising 1 billion particles. Interaction parameters were m = 1, σ₁₁ = 1, σ₁₂ = 0.8, σ₂₂ = 0.88, ε₁₁ = 1, ε₁₂ = 1.5, ε₂₂ = 0.5, with density ρ = 1.2 and initial temperature T = 0.5 (liquid state). The system was cooled slowly from T = 2 down to T = 10⁻⁴. Structural observables (radial distribution function, static structure factor) and dynamical quantities (mean‑square displacement, intermediate scattering function) displayed the characteristic slowdown and dynamical arrest associated with the liquid‑to‑glass transition. Importantly, this transition was captured at a spatial scale two orders of magnitude larger than any previously reported glass‑forming MD simulation, providing unprecedented resolution of heterogeneous dynamics in the micron‑scale regime.
Conclusions: The BBMD package demonstrates that, with careful algorithmic choices, memory‑efficient data structures, and architecture‑specific low‑level optimizations, molecular‑dynamics simulations of up to 100 billion particles can be performed with high parallel efficiency on current petascale machines. The achieved per‑step times (sub‑second for a billion particles) open the door to systematic investigations of glassy dynamics, nucleation, and other collective phenomena that were previously inaccessible due to computational limits. The work sets a new benchmark for large‑scale MD and provides a concrete pathway toward exascale atomistic simulations of complex materials.
Comments & Academic Discussion
Loading comments...
Leave a Comment