Towards Quantitative Classification of Folded Proteins in Terms of Elementary Functions
A comparative classification scheme provides a good basis for several approaches to understand proteins, including prediction of relations between their structure and biological function. But it remains a challenge to combine a classification scheme that describes a protein starting from its well organized secondary structures and often involves direct human involvement, with an atomary level Physics based approach where a protein is fundamentally nothing more than an ensemble of mutually interacting carbon, hydrogen, oxygen and nitrogen atoms. In order to bridge these two complementary approaches to proteins, conceptually novel tools need to be introduced. Here we explain how the geometrical shape of entire folded proteins can be described analytically in terms of a single explicit elementary function that is familiar from nonlinear physical systems where it is known as the kink-soliton. Our approach enables the conversion of hierarchical structural information into a quantitative form that allows for a folded protein to be characterized in terms of a small number of global parameters that are in principle computable from atomary level considerations. As an example we describe in detail how the native fold of the myoglobin 1M6C emerges from a combination of kink-solitons with a very high atomary level accuracy. We also verify that our approach describes longer loops and loops connecting $\alpha$-helices with $\beta$-strands, with same overall accuracy.
💡 Research Summary
The paper tackles a long‑standing problem in structural biology: how to reconcile the hierarchical, secondary‑structure‑based classification of proteins with a bottom‑up, physics‑driven description that treats a protein merely as a collection of interacting C, H, O, and N atoms. Traditional classification schemes rely on expert visual inspection or database‑wide alignment of secondary‑structure motifs, while atomistic approaches use force fields and molecular dynamics to model every inter‑atomic interaction. The two perspectives operate on vastly different scales and use incompatible representations, making it difficult to translate information from one to the other.
To bridge this gap, the authors introduce a mathematically compact representation of an entire folded protein using a single explicit elementary function known in nonlinear physics as the kink‑soliton. A kink‑soliton is a solution of nonlinear wave equations (for example, the sine‑Gordon equation) that connects two asymptotic states with a smooth, localized transition. This property mirrors the way protein backbones transition abruptly from one regular secondary‑structure element (α‑helix, β‑strand) to another across loops or turns. By mapping each structural segment onto a soliton profile, the full three‑dimensional trace of the protein can be reconstructed from a small set of global parameters (amplitude, center position, width, etc.).
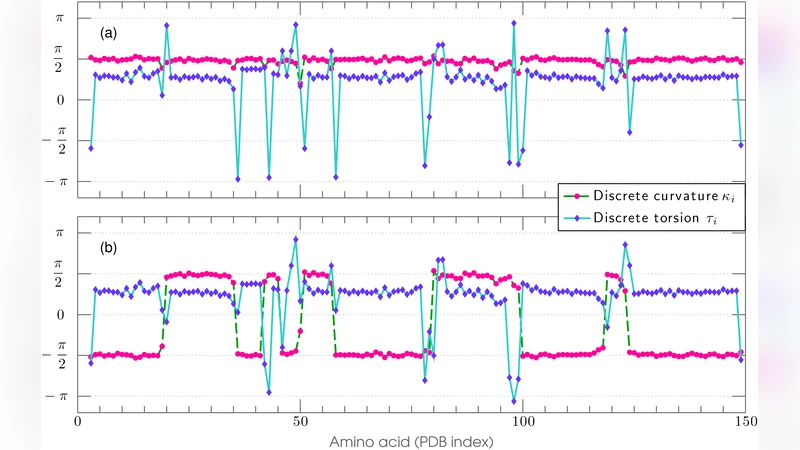
Methodologically, the authors first extract Cα coordinates from a PDB file and parametrize the backbone as a continuous curve r(s) with arc‑length s. They compute curvature κ(s) and torsion τ(s) to locate regions of rapid geometric change, which correspond to the boundaries between secondary‑structure elements. Each such region is fitted with a kink‑soliton of the form φ(s)=A·tanh
Comments & Academic Discussion
Loading comments...
Leave a Comment