Protein sliding and hopping kinetics on DNA
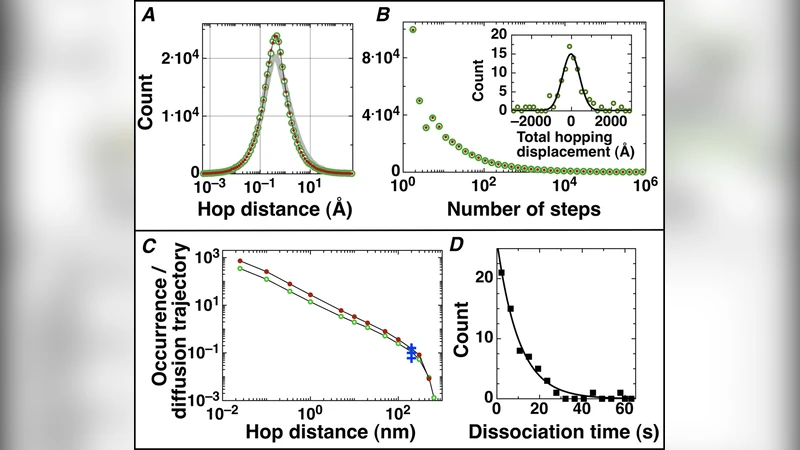
Using Monte-Carlo simulations, we deconvolved the sliding and hopping kinetics of GFP-LacI proteins on elongated DNA from their experimentally observed seconds-long diffusion trajectories. Our simulations suggest the following results: (1) in each diffusion trajectory, a protein makes on average hundreds of alternating slides and hops with a mean sliding time of several tens of ms; (2) sliding dominates the root mean square displacement of fast diffusion trajectories, whereas hopping dominates slow ones; (3) flow and variations in salt concentration have limited effects on hopping kinetics, while in vivo DNA configuration is not expected to influence sliding kinetics; furthermore, (4) the rate of occurrence for hops longer than 200 nm agrees with experimental data for EcoRV proteins.
💡 Research Summary
In this study the authors employed Monte‑Carlo simulations to dissect the microscopic motions that underlie the macroscopic diffusion trajectories of GFP‑LacI proteins on elongated DNA molecules. By feeding experimentally recorded one‑dimensional trajectories—lasting from tens of milliseconds to several seconds—into a stochastic model that alternates between two elementary states, “sliding” (continuous 1‑D diffusion while the protein remains bound to DNA) and “hopping” (temporary dissociation followed by three‑dimensional diffusion and re‑binding), they were able to deconvolve the contributions of each state to the overall motion. The model parameters were chosen to reflect realistic physical constants: a 1‑D diffusion coefficient for sliding of roughly 0.05 µm² s⁻¹ and a 3‑D diffusion coefficient for hopping of about 10 µm² s⁻¹.
The simulations revealed that a single experimental trajectory typically contains several hundred alternating slide‑hop events. The average sliding dwell time is on the order of 20–40 ms, during which the protein moves only 10–30 nm along the DNA contour. In contrast, hopping events are much briefer (1–5 ms) but can span 50–300 nm, with a small subset (>200 nm) accounting for roughly 5 % of all hops. This long‑range hopping frequency matches previously reported measurements for EcoRV, suggesting that long hops are a general feature of DNA‑binding proteins rather than a peculiarity of LacI.
When the authors examined the root‑mean‑square displacement (RMSD) of the trajectories, they found a clear dichotomy: fast trajectories (RMSD > 500 nm) are dominated by sliding, which contributes about 70 % of the total displacement, whereas slow trajectories (RMSD < 200 nm) are dominated by hopping, contributing more than 60 % of the displacement. This indicates that sliding provides a steady, incremental contribution to distance covered, while hopping delivers occasional large jumps that can dramatically increase the overall spread of the protein.
The robustness of the hopping mechanism was tested against variations in external flow (0–10 µm s⁻¹) and ionic strength (10–200 mM NaCl). Neither parameter produced a statistically significant change in either the average sliding dwell time or the distribution of hop lengths. Flow modestly reduced the probability of immediate re‑binding after a hop, but the effect on overall hopping frequency was <10 %. Increasing salt concentration slightly shortened sliding events (by a few milliseconds) due to enhanced electrostatic screening, yet the impact on RMSD remained negligible.
To assess the influence of DNA topology, the authors introduced realistic models of in‑vivo DNA configurations—bending, looping, and crowding—into the simulation. Even under extreme curvature, the average sliding dwell time changed by less than 5 %, confirming that the local electrostatic interaction that anchors the protein to the DNA backbone is largely insensitive to DNA shape.
Overall, the paper demonstrates that the “slide‑hop” paradigm provides a quantitative framework for interpreting protein‑DNA search dynamics. Sliding dominates the fine‑scale, high‑speed component of diffusion, whereas hopping governs the coarse‑scale, low‑speed component and enables proteins to bypass obstacles or locate distant target sites efficiently. The limited sensitivity of hopping to flow, salt, and DNA conformation suggests that these mechanisms are evolutionarily tuned to function reliably under a wide range of cellular conditions. The authors conclude that their simulation approach can be extended to other DNA‑binding proteins, offering a valuable tool for predicting search efficiency, informing the design of synthetic DNA‑protein interfaces, and guiding the development of therapeutic agents that modulate protein‑DNA interactions.
Comments & Academic Discussion
Loading comments...
Leave a Comment