Statistical mechanics of transcription-factor binding site discovery using Hidden Markov Models
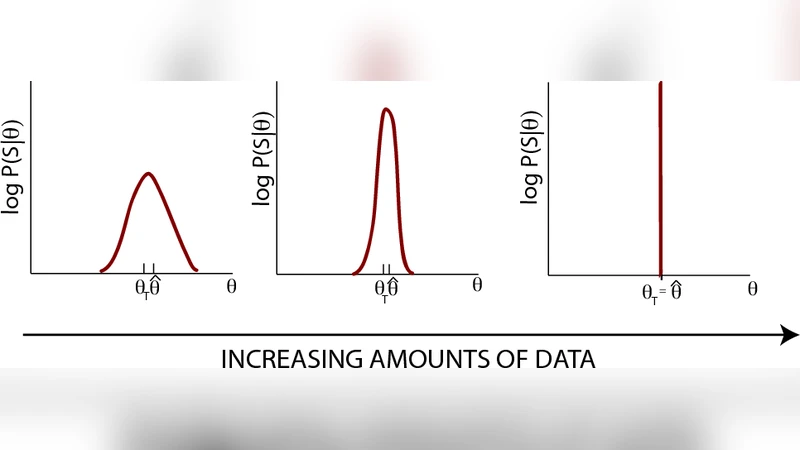
Hidden Markov Models (HMMs) are a commonly used tool for inference of transcription factor (TF) binding sites from DNA sequence data. We exploit the mathematical equivalence between HMMs for TF binding and the “inverse” statistical mechanics of hard rods in a one-dimensional disordered potential to investigate learning in HMMs. We derive analytic expressions for the Fisher information, a commonly employed measure of confidence in learned parameters, in the biologically relevant limit where the density of binding sites is low. We then use techniques from statistical mechanics to derive a scaling principle relating the specificity (binding energy) of a TF to the minimum amount of training data necessary to learn it.
💡 Research Summary
The paper investigates the learning limits of Hidden Markov Models (HMMs) when they are used to discover transcription‑factor (TF) binding sites in DNA sequences. The authors first review the biological problem: TFs bind short DNA motifs (typically 6–20 bp) and the goal of computational methods is to infer a model of these motifs from a limited set of experimentally known sites. Traditional approaches include Position‑Weight Matrices (PWMs), physics‑based algorithms, and various probabilistic models; HMMs are especially popular because they can simultaneously learn a motif (the emission probabilities) and a natural threshold for calling a site (through transition probabilities).
The core contribution of the paper is a rigorous mapping between a TF‑binding HMM and the statistical mechanics of a one‑dimensional gas of hard rods placed in a disordered external potential. In this analogy, each rod represents a binding site of length ℓ, the hard‑core constraint enforces the biological rule that binding sites cannot overlap, and the site‑specific “binding energy” E(S)=ε·S is defined from the emission probabilities via ε_{jα}=−log
Comments & Academic Discussion
Loading comments...
Leave a Comment