Modeling capsid self-assembly: Design and analysis
A series of simulations aimed at elucidating the self-assembly dynamics of spherical virus capsids is described. This little-understood phenomenon is a fascinating example of the complex processes that occur in the simplest of organisms. The fact that different viruses adopt similar structural forms is an indication of a common underlying design, motivating the use of simplified, low-resolution models in exploring the assembly process. Several versions of a molecular dynamics approach are described. Polyhedral shells of different sizes are involved, the assembly pathways are either irreversible or reversible, and an explicit solvent is optionally included. Model design, simulation methodology and analysis techniques are discussed. The analysis focuses on the growth pathways and the nature of the intermediate states, properties that are hard to access experimentally. Among the key observations are that efficient growth proceeds by means of a cascade of highly reversible stages, and that while there are a large variety of possible partial assemblies, only a relatively small number of strongly bonded configurations are actually encountered.
💡 Research Summary
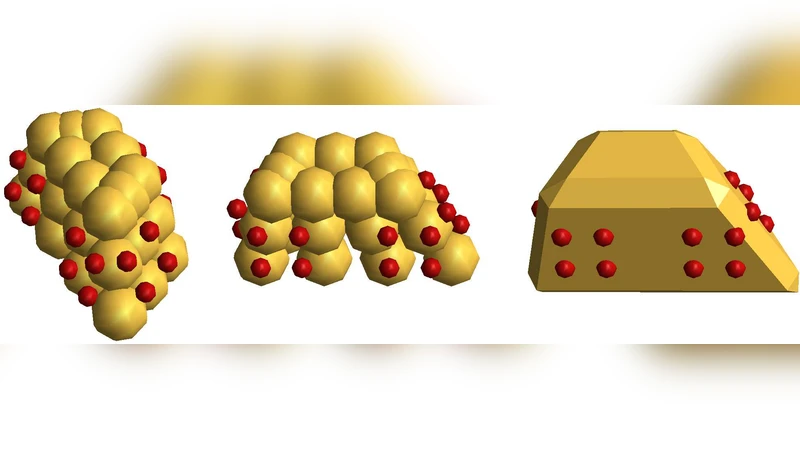
The paper presents a comprehensive study of viral capsid self‑assembly using low‑resolution, coarse‑grained molecular dynamics (MD) simulations. The authors construct simplified capsomer particles from rigid assemblies of soft spheres, each decorated with several attractive sites that interact via an inverse‑power potential smoothly merged into a short‑range harmonic well. Repulsive interactions between the constituent spheres are modeled with a truncated Lennard‑Jones potential to prevent overlap. Two capsid geometries are investigated: a T = 1 icosahedral shell composed of 60 triangular capsomers and a T = 3 shell of 180 trapezoidal capsomers. For the T = 3 case, three particle variants (colored B, G, R) are introduced to implement quasi‑equivalence, allowing slight variations in face angles that mimic the asymmetry observed in real virus capsids.
Four simulation variants are explored: (1) irreversible bonding, where once a bond forms it is permanently locked; (2) reversible bonding, permitting bonds to break and reform, thereby reproducing realistic thermodynamic fluctuations; (3) simulations without explicit solvent, in which particle motion is essentially ballistic and irreversible bonding is used in early stages to reduce computational cost; and (4) simulations with an explicit atomistic solvent, which acts as a heat bath, damps particle trajectories, and enables sub‑assemblies to separate without direct collisions.
The results reveal that efficient capsid formation proceeds through a cascade of highly reversible stages. Small intermediates such as dimers and trimers form rapidly, then repeatedly associate and dissociate, allowing the system to explore many configurations before settling into low‑energy structures. Although combinatorially many partial assemblies are possible, only a limited set of strongly bonded configurations dominate the observed pathways, indicating a strong energetic bias toward specific intermediate geometries. Reversible bonding dramatically increases the yield of complete shells compared with irreversible bonding, because erroneous contacts can be undone and corrected. The inclusion of explicit solvent further enhances reversibility by providing thermal damping and absorbing the exothermic energy released during bond formation.
Particle design details are shown to be critical. Increasing particle thickness and positioning attractive sites within the particle’s geometric envelope reduce unintended contacts and enforce correct face‑to‑face alignment. The quasi‑equivalence scheme for T = 3 capsids demonstrates that modest variations in particle shape can be accommodated without sacrificing overall symmetry, provided that the interaction sites are appropriately matched.
Comparisons with earlier work that employed hard‑sphere, patchy‑particle, or purely spherical models highlight the advantages of the present approach: the extended soft‑sphere architecture combined with multiple, precisely placed interaction sites yields maximal bonding specificity while minimizing spurious aggregation. This design reproduces the high fidelity of natural capsid assembly despite the model’s simplicity.
In summary, the study establishes that low‑resolution, shape‑specific capsomer models coupled with reversible interaction potentials and, when feasible, explicit solvent, can faithfully capture the kinetic and thermodynamic aspects of viral capsid self‑assembly. The findings underscore the importance of reversibility, intermediate‑state energetics, and geometric complementarity in achieving high assembly yields. These insights provide a solid theoretical foundation for the design of synthetic self‑assembling nanostructures, the development of antiviral strategies targeting capsid formation, and future computational investigations of large‑scale biomolecular assembly.
Comments & Academic Discussion
Loading comments...
Leave a Comment