Coarse Graining RNA Nanostructures for Molecular Dynamics Simulations
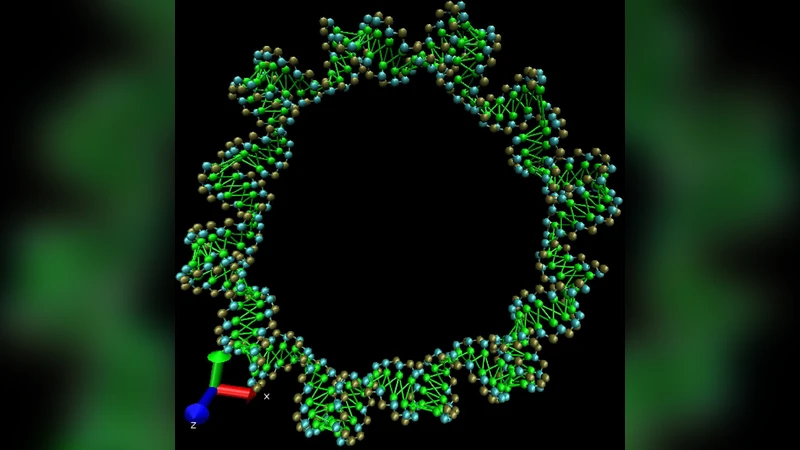
A series of coarse-grained models have been developed for the study of the molecular dynamics of RNA nanostructures. The models in the series have one to three beads per nucleotide and include different amounts of detailed structural information. Such a treatment allows us to reach, for the systems of thousands of nucleotides, a time scale of microseconds (i.e. by three orders of magnitude longer than in the full atomistic modelling) and thus to enable simulations of large RNA polymers in the context of bionanotechnology. We find that the 3-beads-per-nucleotide models, described by a set of just a few universal parameters, are able to describe different RNA conformations and are comparable in structural precision to the models where detailed values of the backbone P-C4’ dihedrals taken from a reference structure are included. These findings are discussed in the context of the RNA conformation classes.
💡 Research Summary
The paper presents a systematic development of coarse‑grained (CG) force fields tailored for RNA nanostructures, aiming to extend molecular dynamics (MD) simulations from the nanosecond regime typical of all‑atom models to the microsecond regime for systems comprising thousands of nucleotides. Three hierarchical CG representations are introduced: a 1‑bead-per‑nucleotide model that captures only the overall shape, a 2‑bead model that separates phosphate and sugar moieties, and a 3‑bead model that additionally resolves the nucleobase. Each bead type is assigned bonded (bond length, angle, dihedral) and non‑bonded (Lennard‑Jones or electrostatic) interactions. The authors distinguish two classes of parameters: (i) a small set of universal parameters that are intended to be transferable across any RNA sequence, and (ii) structure‑specific backbone dihedral values (P‑C4′) extracted from a reference high‑resolution structure.
A key finding is that the 3‑bead model, when parametrized solely with the universal set, reproduces a wide variety of RNA conformations with a structural fidelity comparable to a model that explicitly incorporates the reference dihedral angles. This result is interpreted in the context of RNA “conformation classes,” which posit that backbone dihedrals cluster around a limited number of preferred values. Consequently, a CG model that captures the statistical distribution of these classes can achieve high accuracy without needing detailed, structure‑specific dihedral inputs.
The authors validate their models on several test systems: canonical A‑form helices, internal loops, three‑way junctions, and larger nanostructures such as RNA nanotubes and switch‑like constructs. Metrics include root‑mean‑square deviation (RMSD) relative to experimental X‑ray or cryo‑EM structures, radius of gyration (Rg), and preservation of secondary‑structure elements. The 3‑bead CG model consistently yields RMSD values below 3 Å for systems up to several thousand nucleotides, while allowing time steps on the order of 10–20 fs. This enables simulations of 10⁶–10⁷ steps per day on commodity hardware, translating into microsecond‑scale trajectories that capture slow processes such as self‑assembly, large‑scale bending, and thermal expansion—phenomena inaccessible to all‑atom MD due to prohibitive computational cost.
Beyond methodological development, the paper discusses practical implications for RNA nanotechnology. The ability to simulate large RNA assemblies over biologically relevant timescales opens avenues for rational design of RNA‑based nanomachines, drug‑delivery vehicles, and responsive switches. The universal parameter set simplifies the workflow: a user can construct a CG model for a novel sequence without performing extensive parameter fitting, then run long‑time MD to assess stability, flexibility, and functional motions. The authors also outline a prospective multiscale framework where CG simulations guide the selection of regions for subsequent all‑atom refinement, thereby combining the speed of CG with the detailed insight of atomistic models.
In summary, this work demonstrates that a modestly detailed 3‑bead CG representation, equipped with a small, transferable parameter library, can faithfully reproduce the structural and dynamic properties of diverse RNA nanostructures on microsecond timescales. The approach balances computational efficiency with sufficient chemical realism, making it a valuable tool for both fundamental studies of RNA physics and the engineering of RNA‑based nanodevices.
Comments & Academic Discussion
Loading comments...
Leave a Comment