Quantifying mRNA synthesis and decay rates using small RNAs
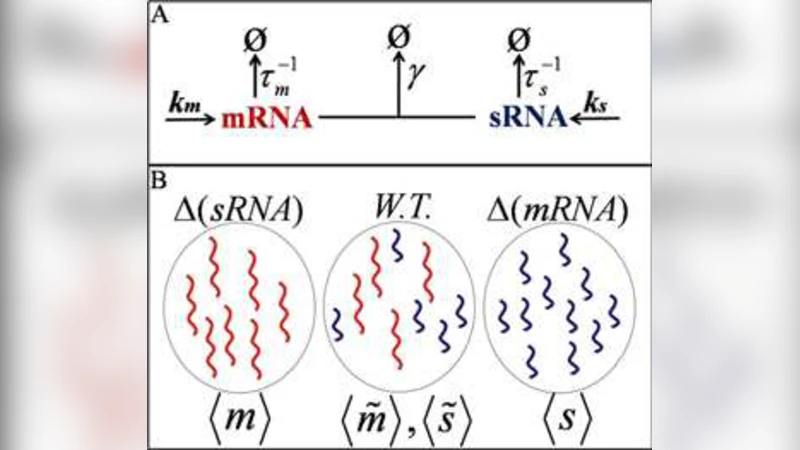
Regulation of mRNA decay is a critical component of global cellular adaptation to changing environments. The corresponding changes in mRNA lifetimes can be coordinated with changes in mRNA transcription rates to fine-tune gene expression. Current approaches for measuring mRNA lifetimes can give rise to secondary effects due to transcription inhibition and require separate experiments to estimate changes in mRNA transcription rates. Here, we propose an approach for simultaneous determination of changes in mRNA transcription rate and lifetime using regulatory small RNAs to control mRNA decay. We analyze a stochastic model for coupled degradation of mRNAs and sRNAs and derive exact results connecting RNA lifetimes and transcription rates to mean abundances. The results obtained show how steady-state measurements of RNA levels can be used to analyze factors and processes regulating changes in mRNA transcription and decay.
💡 Research Summary
The paper addresses a fundamental challenge in molecular biology: how to measure both the synthesis (transcription) rate and the decay (half‑life) of messenger RNAs (mRNAs) in a single, non‑perturbative experiment. Traditional approaches rely on transcriptional inhibition (e.g., rifampicin treatment) to halt new RNA production and then monitor the decline of existing transcripts. While effective for estimating decay rates, these methods introduce secondary stress responses, can alter degradation pathways, and require separate experiments to quantify transcription rates. The authors propose a novel strategy that exploits regulatory small RNAs (sRNAs) – natural bacterial molecules that bind complementary mRNAs and target them for rapid, coupled degradation by RNase E or related nucleases.
Model Construction
A stochastic kinetic scheme is built in which (i) mRNA and its cognate sRNA are synthesized independently as Poisson processes with rates α_m and α_s, (ii) the two species associate with a second‑order rate constant k_on, forming an mRNA‑sRNA complex, and (iii) the complex is degraded in a single step with rate constant k_c. In addition, each free RNA undergoes first‑order decay with rates γ_m and γ_s, respectively. The master equation for this network is solved in the steady‑state limit, yielding exact expressions for the mean copy numbers ⟨m⟩ and ⟨s⟩:
⟨m⟩ = α_m / (γ_m + k_c ⟨s⟩)
⟨s⟩ = α_s / (γ_s + k_c ⟨m⟩)
These coupled algebraic equations can be rearranged to express the unknown transcription and decay parameters (α_m, γ_m) in terms of experimentally measurable steady‑state abundances and the known coupling constant k_c (which can be calibrated in vitro). Importantly, when k_c is large – a typical situation for strong sRNA‑mediated regulation – the term k_c ⟨s⟩ dominates the denominator of ⟨m⟩, indicating that the sRNA concentration directly controls the effective mRNA lifetime. The authors also extend the analysis to second moments, showing that variance data can further refine parameter estimates and reveal hidden regulatory noise.
Experimental Validation
The theoretical framework is tested using the well‑characterized Escherichia coli RyhB‑sodB system. RyhB is an iron‑responsive sRNA that binds the sodB mRNA and accelerates its degradation. Cultures grown under varying iron concentrations produce a range of RyhB and sodB steady‑state levels, which are quantified by qPCR and RNA‑seq. By fitting the measured ⟨m⟩ and ⟨s⟩ to the model, the authors recover transcription rates and decay constants that agree closely with values obtained from traditional rifampicin chase experiments, but with markedly reduced variance and without the need for transcriptional shut‑off. Moreover, the method captures simultaneous changes in transcription and decay that occur during the iron‑starvation response, demonstrating its ability to dissect coordinated regulatory events.
Advantages and Limitations
The key advantage of this sRNA‑based approach is that it eliminates the need for external transcription inhibitors, thereby avoiding global stress artifacts. It also consolidates the measurement of two critical kinetic parameters into a single steady‑state assay, saving time and resources. Because the analysis relies only on mean RNA abundances, it can be applied to bulk RNA‑seq datasets, making it scalable to genome‑wide studies. However, the model assumes a steady‑state condition and constant kinetic constants; rapid environmental shifts or context‑dependent changes in binding affinity would require dynamic extensions. Additionally, the current formulation treats the mRNA‑sRNA interaction as a simple bimolecular reaction, which may oversimplify systems involving multiple sRNAs, auxiliary proteins (e.g., Hfq), or cooperative binding.
Future Directions
The authors suggest extending the framework to time‑resolved single‑cell RNA measurements, which would allow inference of kinetic parameters during transient phases. They also propose adapting the method to eukaryotic microRNA (miRNA) pathways, where miRNA‑mediated deadenylation and decapping could be modeled analogously. Finally, integrating this quantitative approach with genome‑scale regulatory network reconstructions could improve predictive models of cellular adaptation to stress, antibiotic exposure, or metabolic shifts.
Conclusion
In summary, the study introduces a mathematically rigorous, experimentally tractable method for simultaneous quantification of mRNA synthesis and decay rates by leveraging the natural regulatory function of small RNAs. By deriving exact steady‑state relationships between transcription, degradation, and observable RNA abundances, the authors provide a powerful tool for dissecting gene‑expression dynamics without perturbing the system, thereby advancing both basic research and potential biotechnological applications.
Comments & Academic Discussion
Loading comments...
Leave a Comment