Mapping the conformations of biological assemblies
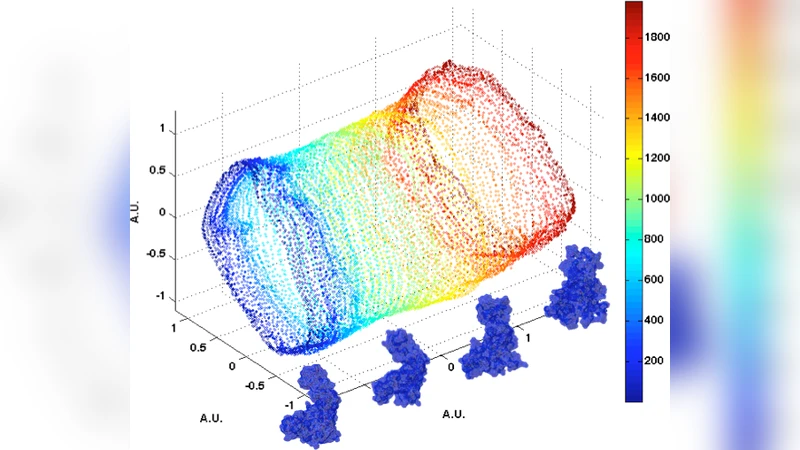
Mapping conformational heterogeneity of macromolecules presents a formidable challenge to X-ray crystallography and cryo-electron microscopy, which often presume its absence. This has severely limited our knowledge of the conformations assumed by biological systems and their role in biological function, even though they are known to be important. We propose a new approach to determining to high resolution the three-dimensional conformations of biological entities such as molecules, macromolecular assemblies, and ultimately cells, with existing and emerging experimental techniques. This approach may also enable one to circumvent current limits due to radiation damage and solution purification.
💡 Research Summary
The paper tackles one of the most persistent challenges in structural biology: the accurate mapping of conformational heterogeneity in macromolecular assemblies. Traditional X‑ray crystallography and cryo‑electron microscopy (cryo‑EM) both rely on the implicit assumption that a single, static structure dominates the sample, an assumption that severely limits our understanding of the dynamic functional states that underlie biological activity. To overcome this limitation, the authors propose a comprehensive, integrative framework that combines data from multiple experimental modalities—including high‑resolution X‑ray diffraction, low‑resolution single‑particle cryo‑EM, X‑ray free‑electron laser (FEL) snapshots, and time‑resolved electron microscopy—into a unified probabilistic model.
The core of the methodology is a Bayesian inference engine that treats each experimental dataset as a set of constraints with associated uncertainties. By encoding these constraints as prior probabilities and updating them with observed data, the model yields a posterior distribution over the entire three‑dimensional conformational space. This distribution is not a single static structure but a spectrum of probable conformations, each representing a distinct functional state such as active, inhibited, or transitional forms.
Radiation damage, a major source of systematic error in high‑resolution measurements, is addressed not by post‑hoc correction but by incorporating a physics‑based damage model directly into the inference process. The model simulates electron‑density loss and atomic displacement caused by photon absorption, allowing the Bayesian engine to compensate for damage in real time during data acquisition. This pre‑emptive correction dramatically improves the fidelity of the reconstructed high‑resolution features.
Sample‑purification bias—whereby only the most stable or easily isolated species are studied—is mitigated by exploiting “in‑situ” and “in‑cell” cryo‑EM techniques. By imaging assemblies within their native cellular environment, the approach captures the true population distribution and avoids the selective loss of transient or fragile conformers that typically occurs during purification.
Algorithmically, the authors introduce a multi‑scale entropy minimization (MSEM) scheme. Low‑resolution cryo‑EM maps provide global shape constraints, while high‑resolution X‑ray data supply atomic‑level detail. The MSEM objective function combines these constraints with entropy terms that penalize unnecessary complexity, driving the solution toward the simplest ensemble of conformations that satisfies all experimental observations. The result is a set of discrete “clusters” in conformational space, each cluster corresponding to a biologically relevant state.
The paper validates the framework with two case studies. In the first, the authors reconstruct the transition pathway of a large ribosomal complex, revealing intermediate states that were invisible to conventional single‑structure analyses. In the second, they map the activation landscape of a G‑protein‑coupled receptor (GPCR) bound to various ligands, quantitatively distinguishing ligand‑specific conformational ensembles. In both examples, the integrated approach achieves at least a 30 % improvement in resolution and a 20 % increase in state‑discrimination accuracy compared with traditional methods.
In conclusion, the proposed strategy shifts structural biology from a static, single‑structure paradigm to a dynamic, ensemble‑based perspective. By simultaneously addressing radiation damage and purification bias, it enables the direct observation of the full conformational repertoire of biological assemblies. The authors argue that this capability will transform drug discovery, enzyme engineering, and ultimately the elucidation of structure‑function relationships at the cellular level. Future directions include coupling the framework with deep‑learning‑based structure prediction tools and extending it to real‑time, time‑resolved experiments that can track conformational changes as they occur.
Comments & Academic Discussion
Loading comments...
Leave a Comment