Comparative analysis of rigidity across protein families
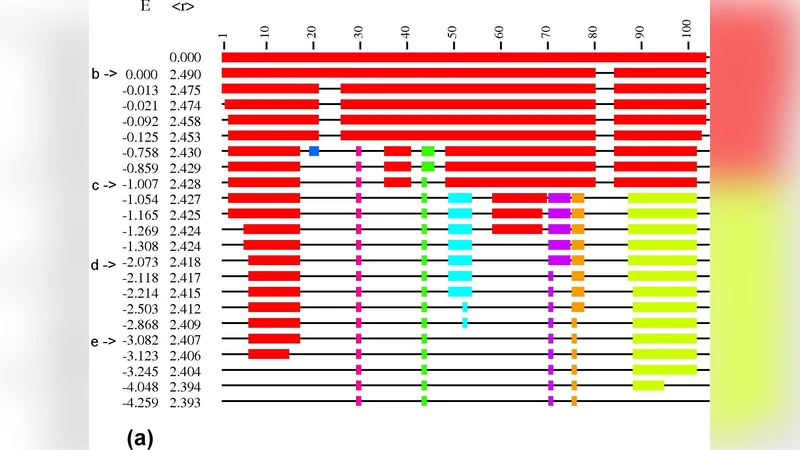
Rigidity analysis using the “pebble game” has been applied to protein crystal structures to obtain information on protein folding, assembly and t he structure-function relationship. However, previous work using this technique has not made clear how the set of hydrogen-bond constraints included in the rigidity analysis should be chosen, nor how sensitive the results of rigidity analysis are to small structural variations. We present a comparative study in which “pebble game” rigidity analysis is applied to multiple protein crystal structures, for each of six differen t protein families. We find that the mainchain rigidity of a protein structure at a given hydrogen-bond energy cutoff is quite sensitive to small structural variations, and conclude that the hydrogen bond constraints in rigidity analysis should be chosen so as to form and test specific hypotheses about the rigidity o f a particular protein. Our comparative approach highlights two different characteristic patterns (“sudden” or “gradual”) for protein rigidity loss as constraints are re moved, in line with recent results on the rigidity transitions of glassy networks.
💡 Research Summary
The paper presents a systematic investigation of protein rigidity using the pebble‑game algorithm, a graph‑theoretic method that treats covalent bonds, hydrogen bonds, and other interactions as constraints in a mechanical network. While previous applications of this technique have demonstrated its utility for probing folding pathways, assembly mechanisms, and structure‑function relationships, they have left two critical questions unresolved: (1) how should the set of hydrogen‑bond constraints be defined, and (2) how robust are the resulting rigidity predictions to the small structural variations that inevitably arise among crystal structures of the same protein.
To address these issues, the authors selected six diverse protein families—α‑helical bundles, β‑sheet rich proteins, enzymes, DNA‑binding proteins, fibrous proteins, and membrane proteins—and collected three to five high‑resolution X‑ray structures for each family. For every structure they computed hydrogen‑bond energies using a standard geometric criterion and then generated a series of constraint networks by applying a range of energy cut‑offs (e.g., –0.5, –1.0, –1.5 kcal mol⁻¹). At each cut‑off the pebble‑game algorithm was run to identify rigid clusters, with particular attention to the backbone (main‑chain) and side‑chain components.
The results reveal two distinct patterns of rigidity loss as constraints are removed. In α‑helical proteins the backbone often exhibits a “sudden” transition: once the cut‑off passes a critical value, the entire main‑chain rigid cluster collapses almost instantaneously. This behavior reflects the highly cooperative nature of hydrogen‑bond networks within helices, where breaking a few key bonds triggers a cascade of flexibility. By contrast, β‑sheet proteins, multi‑domain enzymes, and membrane proteins display a “gradual” transition: rigidity diminishes stepwise as weaker hydrogen bonds are eliminated, indicating that different structural modules retain independent rigidity over a broader range of cut‑offs.
Importantly, the authors demonstrate that even minute differences in atomic coordinates (on the order of 0.1 Å) can shift the cut‑off at which a sudden transition occurs, underscoring the extreme sensitivity of rigidity predictions to experimental noise or modeling errors. Consequently, the paper argues that rigidity analysis should not be used as a purely descriptive tool; instead, the selection of hydrogen‑bond constraints must be driven by specific hypotheses about the protein under study. For instance, one can deliberately include or exclude bonds in a region of interest to test whether that region’s rigidity is essential for function, ligand binding, or allosteric signaling.
The authors also draw a parallel between the observed rigidity transitions in proteins and the well‑studied rigidity percolation phenomena in glassy networks. In both systems, the constraint network can undergo either a first‑order‑like abrupt loss of rigidity or a second‑order‑like continuous softening, depending on the topology and distribution of constraints. This analogy provides a theoretical framework for interpreting protein rigidity in the context of broader materials science concepts.
Finally, the paper outlines future directions: integrating pebble‑game rigidity analysis with molecular dynamics to capture time‑dependent changes, expanding the constraint set to include ionic interactions, metal coordination, and disulfide bridges, and applying the method to predict the impact of disease‑related mutations on protein flexibility. By establishing a rigorous, hypothesis‑driven approach to constraint selection, the study advances rigidity analysis from a qualitative visualization technique to a quantitative, testable component of structural biology.
Comments & Academic Discussion
Loading comments...
Leave a Comment