Path integral evaluation of equilibrium isotope effects
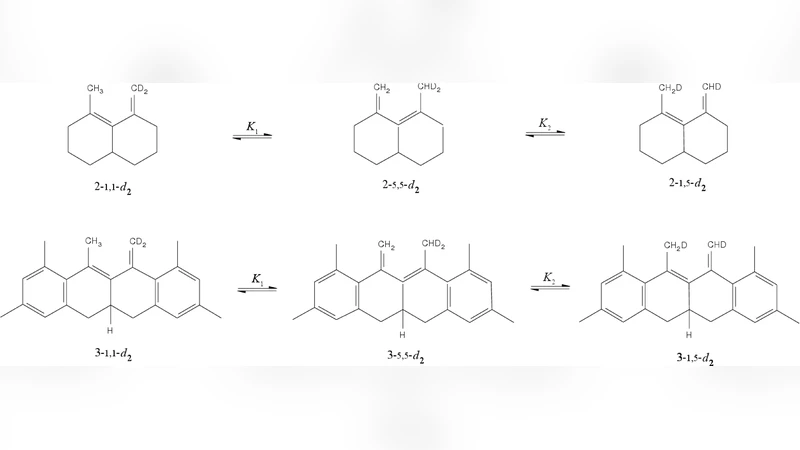
A general and rigorous methodology to compute the quantum equilibrium isotope effect is described. Unlike standard approaches, ours does not assume separability of rotational and vibrational motions and does not make the harmonic approximation for vibrations or rigid rotor approximation for the rotations. In particular, zero point energy and anharmonicity effects are described correctly quantum mechanically. The approach is based on the thermodynamic integration with respect to the mass of isotopes and on the Feynman path integral representation of the partition function. An efficient estimator for the derivative of free energy is used whose statistical error is independent of the number of imaginary time slices in the path integral, speeding up calculations by a factor of 60 at 500 K. We describe the implementation of the methodology in the molecular dynamics package Amber 10. The method is tested on three [1,5] sigmatropic hydrogen shift reactions. Because of the computational expense, we use ab initio potentials to evaluate the equilibrium isotope effects within the harmonic approximation, and then the path integral method together with semiempirical potentials to evaluate the anharmonicity corrections. Our calculations show that the anharmonicity effects amount up to 30% of the symmetry reduced reaction free energy. The numerical results are compared with recent experiments of Doering and coworkers, confirming the accuracy of the most recent measurement on 2,4,6,7,9-pentamethyl-5-(5,5-$^2$H$_2$)methylene-11,11a-dihydro-12H-naphthacene as well as concerns about compromised accuracy, due to side reactions, of another measurement on 2-methyl-10-(10,10-$^2$H$_2$)methylenebicyclo[4.4.0]dec-1-ene.
💡 Research Summary
The paper presents a rigorous and general methodology for calculating quantum equilibrium isotope effects (EIEs) without relying on the usual separability of rotational and vibrational motions or on the harmonic and rigid‑rotor approximations. The authors combine thermodynamic integration (TI) with respect to the isotopic mass and the Feynman path‑integral (PI) representation of the canonical partition function. The central technical advance is an estimator for the derivative of the free energy with respect to mass whose statistical error does not grow with the number of imaginary‑time slices (beads) used in the PI discretization. This “bead‑independent” estimator enables a dramatic speed‑up: at 500 K the new approach converges about 60 times faster than conventional primitive‑estimator PI‑MD.
Implementation was carried out within the Amber 10 molecular‑dynamics package. The authors adopt a two‑stage strategy to keep the computational cost manageable. First, high‑level ab initio electronic‑structure methods (e.g., MP2, B3LYP‑D3) are used to evaluate the EIE under the harmonic approximation, providing a baseline free‑energy difference. Second, semi‑empirical potentials (PM6, AM1) are employed together with the bead‑independent PI‑MD to compute anharmonic corrections. By adding these corrections to the harmonic baseline, the full quantum EIE—including zero‑point energy, tunnelling, and anharmonicity—is obtained with a modest computational expense.
The methodology was tested on three prototypical