Tight-binding modeling of charge migration in DNA devices
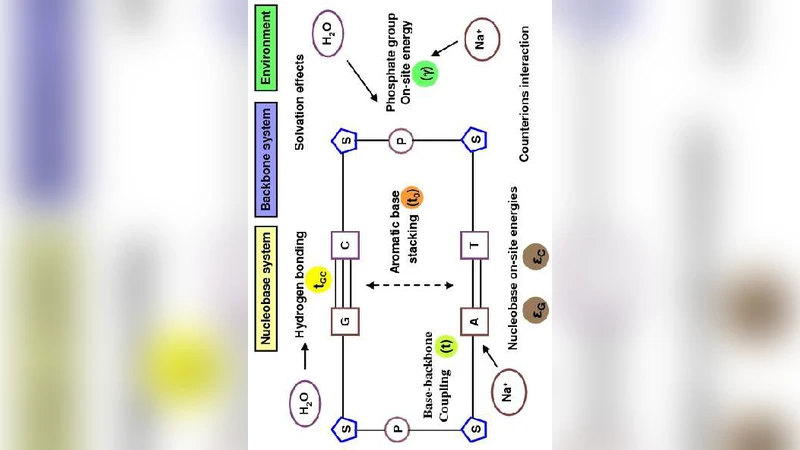
Long range charge transfer experiments in DNA oligomers and the subsequently measured – and very diverse – transport response of DNA wires in solid state experiments exemplifies the need for a thorough theoretical understanding of charge migration in DNA-based natural and artificial materials. Here we present a review of tight-binding models for DNA conduction which have the intrinsic merit of containing more structural information than plain rate-equation models while still retaining sufficient detail of the electronic properties. This allows for simulations of transport properties to be more manageable with respect to density functional theory methods or correlated first principle algorithms.
💡 Research Summary
The paper provides a comprehensive review of tight‑binding (TB) models as a middle‑ground theoretical framework for describing charge migration in DNA‑based materials. It begins by highlighting the experimental paradox that DNA oligomers can support long‑range charge transfer in solution, while solid‑state DNA wires exhibit a wide spectrum of transport behaviors ranging from metallic to insulating. Traditional rate‑equation approaches, which treat charge hopping as a sequence of stochastic events with averaged rates, fail to capture the influence of base‑pair sequence, structural fluctuations, and electrode‑DNA coupling on the electronic properties.
The authors then introduce the TB formalism, where each nucleotide (or base‑pair) is represented by a single orbital with an on‑site energy ε and nearest‑neighbor hopping integrals t that encode π‑π stacking interactions. These parameters are not static; they depend on the helical geometry (twist angle, rise per base), solvent screening, ionic strength, and local disorder such as mismatches or chemical modifications. By calibrating ε and t against density‑functional theory (DFT) calculations or spectroscopic data, the TB Hamiltonian can retain essential chemical specificity while remaining computationally tractable.
A major strength of the TB approach is its compatibility with nonequilibrium Green’s function (NEGF) and transfer‑matrix techniques, which allow the direct calculation of I‑V characteristics, transmission spectra, and length‑dependent conductance for realistic device geometries. The paper details how to embed the TB Hamiltonian within an electrode‑DNA‑electrode setup, accounting for contact self‑energies, voltage drops, and possible charge redistribution at the interfaces. By varying the TB parameters, the authors reproduce experimentally observed trends: guanine‑rich sequences, which have lower ionization potentials, yield larger t values and higher conductance, whereas adenine‑thymine rich regions act as tunneling barriers.
The review also discusses extensions that incorporate electron‑phonon coupling, typically via Holstein‑type or Su‑Schrieffer‑Heeger‑type models, to capture temperature‑dependent dephasing and polaron formation. These extensions explain why charge transport can shift from coherent tunneling in short oligomers to thermally activated hopping in longer wires. Moreover, the authors examine artificial DNA nanowires, such as metal‑decorated strands or DNA‑graphene hybrids, showing how the TB framework can be adapted to include additional hopping pathways or substrate‑induced band renormalization.
Finally, the paper acknowledges current limitations: TB models generally neglect strong electron correlation and many‑body effects, which may be important in highly charged or strongly coupled systems. The authors suggest integrating Hubbard‑type on‑site interactions or employing GW‑based corrections to improve accuracy. They also call for multiscale simulations that couple TB electronic structure with molecular dynamics of the surrounding solvent and counter‑ions, enabling a realistic description of dynamic disorder.
In conclusion, tight‑binding models strike a balance between the atomistic detail of first‑principles methods and the simplicity of phenomenological rate equations. They provide a versatile platform for exploring charge migration in both natural DNA and engineered DNA‑based nanodevices, facilitating quantitative comparison with experiment and guiding the design of future molecular electronics.
Comments & Academic Discussion
Loading comments...
Leave a Comment