A first-principles model of early evolution: Emergence of gene families, species and preferred protein folds
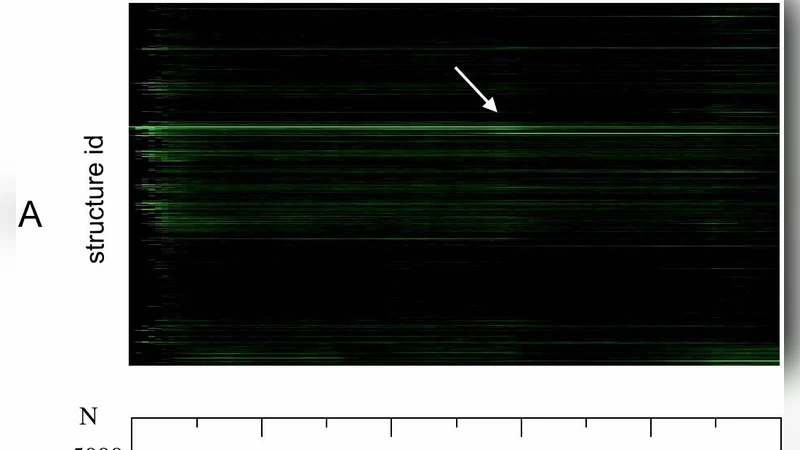
In this work we develop a microscopic physical model of early evolution, where phenotype,organism life expectancy, is directly related to genotype, the stability of its proteins in their native conformations which can be determined exactly in the model. Simulating the model on a computer, we consistently observe the Big Bang scenario whereby exponential population growth ensues as soon as favorable sequence-structure combinations (precursors of stable proteins) are discovered. Upon that, random diversity of the structural space abruptly collapses into a small set of preferred proteins. We observe that protein folds remain stable and abundant in the population at time scales much greater than mutation or organism lifetime, and the distribution of the lifetimes of dominant folds in a population approximately follows a power law. The separation of evolutionary time scales between discovery of new folds and generation of new sequences gives rise to emergence of protein families and superfamilies whose sizes are power-law distributed, closely matching the same distributions for real proteins. On the population level we observe emergence of species, subpopulations which carry similar genomes. Further we present a simple theory that relates stability of evolving proteins to the sizes of emerging genomes. Together, these results provide a microscopic first principles picture of how first gene families developed in the course of early evolution
💡 Research Summary
The paper presents a first‑principles, microscopic model of early biological evolution in which an organism’s phenotype—its life expectancy—is directly linked to the thermodynamic stability of its encoded proteins. Using a lattice‑protein framework (27‑residue sequences on a 3 × 3 grid) and the Miyazawa‑Jernigan contact potential, the authors can calculate the native‑state free energy (ΔG) of every possible fold exactly. A protein is deemed stable when ΔG < 0, and an organism’s survival probability is a monotonic function of the average stability of its proteome. This explicit genotype‑to‑phenotype mapping allows the authors to simulate populations of replicating organisms subject to point mutations, while tracking both population size and the distribution of protein structures over evolutionary time.
The simulations reveal two distinct dynamical regimes. In the first, termed the “Big Bang” scenario, the population remains near‑stagnant until a rare sequence‑structure combination that yields a sufficiently stable protein appears. This “precursor” dramatically raises the replication rate, leading to exponential population growth and a rapid increase in genetic diversity. Once the population has expanded, the second regime emerges: the structural space collapses from the full repertoire of over 100 000 possible folds to a handful of “preferred” folds. These dominant folds have lifetimes far exceeding the typical mutation time or organism lifespan, and the distribution of their persistence times follows a power‑law, indicating that rare, large‑scale events shape the long‑term architecture of the protein universe.
After a preferred fold becomes established, the model generates a large number of sequence variants that share the same native structure. These variants constitute protein families and, at higher hierarchical levels, superfamilies. The size distribution of families and superfamilies also follows a power‑law, with exponents that closely match those observed in real protein databases such as Pfam and SCOP. Thus, the model reproduces the empirical scaling laws of protein evolution without invoking any ad‑hoc assumptions; they arise naturally from the interplay between stability selection and mutation.
On the population level, groups of organisms that share the same dominant fold and a similar set of genes begin to cluster together. The authors refer to these clusters as nascent “species.” Gene flow between clusters diminishes over time, leading to clear genetic boundaries reminiscent of speciation events. This emergent speciation demonstrates that even in a minimalist physical model, the separation of evolutionary time scales—fast sequence drift versus slow discovery of new folds—can generate higher‑order biological organization.
To rationalize the observed relationship between protein stability and genome size, the authors develop a simple analytical theory. Stable proteins increase the organism’s replication efficiency, allowing the genome to expand, but a larger genome also raises the mutational load, which in turn imposes stronger selective pressure for stability. The balance between these opposing forces yields a “stability‑capacity curve” that predicts a maximal viable genome size for a given average protein stability. Simulation data fit this curve remarkably well, providing a quantitative link between molecular thermodynamics and macro‑evolutionary trends.
In summary, the study offers a comprehensive, physics‑based picture of early evolution. By coupling exact protein folding energetics to organismal fitness, it demonstrates how exponential population growth, collapse of structural diversity into a few robust folds, power‑law distributions of protein families, and the emergence of species can all arise from first principles. The model bridges the gap between microscopic molecular constraints and macroscopic evolutionary patterns, offering valuable insights for both origin‑of‑life research and contemporary studies of protein evolution.
Comments & Academic Discussion
Loading comments...
Leave a Comment