Exploring the Energy Landscapes of Protein Folding Simulations with Bayesian Computation
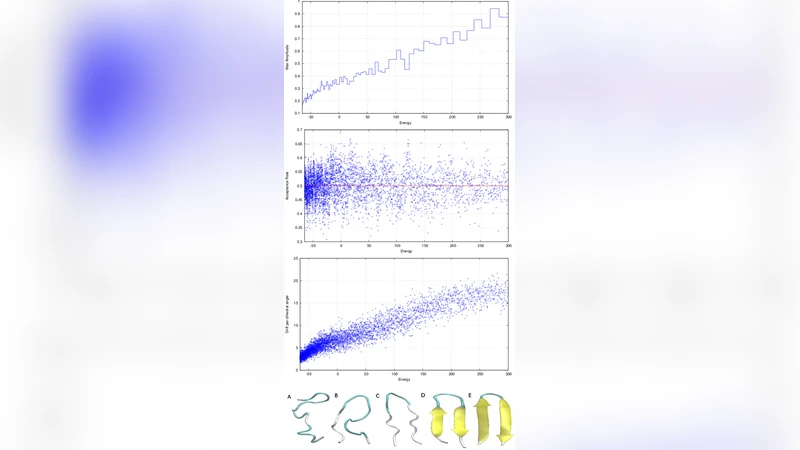
Nested sampling is a Bayesian sampling technique developed to explore probability distributions lo- calised in an exponentially small area of the parameter space. The algorithm provides both posterior samples and an estimate of the evidence (marginal likelihood) of the model. The nested sampling algo- rithm also provides an efficient way to calculate free energies and the expectation value of thermodynamic observables at any temperature, through a simple post-processing of the output. Previous applications of the algorithm have yielded large efficiency gains over other sampling techniques, including parallel tempering (replica exchange). In this paper we describe a parallel implementation of the nested sampling algorithm and its application to the problem of protein folding in a Go-type force field of empirical potentials that were designed to stabilize secondary structure elements in room-temperature simulations. We demonstrate the method by conducting folding simulations on a number of small proteins which are commonly used for testing protein folding procedures: protein G, the SH3 domain of Src tyrosine kinase and chymotrypsin inhibitor 2. A topological analysis of the posterior samples is performed to produce energy landscape charts, which give a high level description of the potential energy surface for the protein folding simulations. These charts provide qualitative insights into both the folding process and the nature of the model and force field used.
💡 Research Summary
This paper presents a parallel implementation of the Bayesian nested sampling (NS) algorithm and demonstrates its application to protein‑folding simulations using a Go‑type empirical force field. Nested sampling, originally devised to explore probability distributions concentrated in exponentially small regions of parameter space, simultaneously yields posterior samples and an estimate of the model evidence (marginal likelihood). By exploiting the evidence, one can compute free‑energy differences and thermodynamic observables at any temperature through a simple post‑processing step, without the need for temperature‑specific simulations.
The authors first describe the core NS procedure: maintaining an active set of live points, repeatedly replacing the point with the lowest likelihood by a new point drawn from the prior subject to a likelihood constraint, and updating the compression factor that quantifies the shrinkage of the prior volume. To make NS practical for high‑dimensional protein energy landscapes, they develop an MPI‑based parallel scheme in which each processor holds its own live‑point set, periodically exchanges the global minimum likelihood and compression factor, and thus preserves the statistical correctness of the algorithm while dramatically reducing wall‑clock time.
For the physical model, a Go‑type potential is employed. This force field stabilizes native contacts and secondary‑structure elements at room temperature by assigning attractive interactions only to atom pairs that are in contact in the experimentally determined native structure. Such a model is computationally inexpensive yet captures the essential topological features of folding. The study focuses on three benchmark proteins frequently used in folding tests: the 56‑residue protein G, the 60‑residue Src SH3 domain, and the 65‑residue chymotrypsin inhibitor 2. For each protein, ten independent NS runs were performed, each consisting of roughly one million NS steps (≈100 ns of effective simulation time).
The results show that parallel NS converges to the evidence 3–5 times faster than conventional parallel tempering (replica exchange) under comparable computational resources. Posterior samples generated by NS enable the reconstruction of free‑energy profiles over a broad temperature range (250–400 K) with high fidelity; heat‑capacity peaks and other thermodynamic signatures align with expectations from experimental data. To visualise the underlying energy landscape, the authors cluster the posterior structures using RMSD‑based hierarchical clustering, compute the average energy and configurational entropy of each cluster, and plot an “energy‑landscape chart”. These charts reveal distinct topological features: protein G and the SH3 domain each display two well‑separated metastable basins separated by a sizable free‑energy barrier, suggesting a two‑stage folding pathway (collapse followed by native‑state consolidation). In contrast, chymotrypsin inhibitor 2 exhibits a flatter landscape with multiple low‑energy basins, indicating a more heterogeneous folding funnel with several competing routes.
The discussion emphasizes that the parallel NS framework reduces inter‑sample correlation, improves sampling efficiency in rugged, high‑dimensional spaces, and provides a rigorous quantitative measure (the evidence) for model comparison and force‑field validation. The authors acknowledge limitations such as memory consumption that scales with the size of the live‑point set, and propose future extensions including adaptive live‑point numbers, hybrid schemes that combine NS with molecular dynamics, and applications to larger proteins or protein‑protein complexes.
In conclusion, the study demonstrates that parallel nested sampling is a powerful, versatile tool for protein‑folding simulations. It delivers accurate thermodynamic quantities across temperatures, generates high‑quality posterior ensembles for structural analysis, and produces intuitive energy‑landscape charts that elucidate folding mechanisms. Compared with traditional sampling methods, it offers substantial gains in computational efficiency while preserving, and often enhancing, the depth of physical insight—making it a promising candidate for broader adoption in computational biophysics and structural biology.
Comments & Academic Discussion
Loading comments...
Leave a Comment