3D Protein Structure Predicted from Sequence
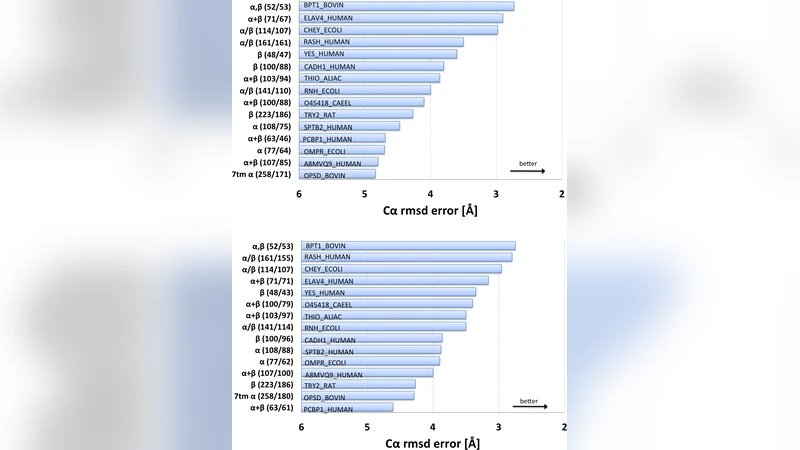
The evolutionary trajectory of a protein through sequence space is constrained by function and three-dimensional (3D) structure. Residues in spatial proximity tend to co-evolve, yet attempts to invert the evolutionary record to identify these constraints and use them to computationally fold proteins have so far been unsuccessful. Here, we show that co-variation of residue pairs, observed in a large protein family, provides sufficient information to determine 3D protein structure. Using a data-constrained maximum entropy model of the multiple sequence alignment, we identify pairs of statistically coupled residue positions which are expected to be close in the protein fold, termed contacts inferred from evolutionary information (EICs). To assess the amount of information about the protein fold contained in these coupled pairs, we evaluate the accuracy of predicted 3D structures for proteins of 50-260 residues, from 15 diverse protein families, including a G-protein coupled receptor. These structure predictions are de novo, i.e., they do not use homology modeling or sequence-similar fragments from known structures. The resulting low C{\alpha}-RMSD error range of 2.7-5.1{\AA}, over at least 75% of the protein, indicates the potential for predicting essentially correct 3D structures for the thousands of protein families that have no known structure, provided they include a sufficiently large number of divergent sample sequences. With the current enormous growth in sequence information based on new sequencing technology, this opens the door to a comprehensive survey of protein 3D structures, including many not currently accessible to the experimental methods of structural genomics. This advance has potential applications in many biological contexts, such as synthetic biology, identification of functional sites in proteins and interpretation of the functional impact of genetic variants.
💡 Research Summary
The paper “3D Protein Structure Predicted from Sequence” presents a groundbreaking approach that leverages evolutionary information encoded in multiple‑sequence alignments (MSAs) to predict protein three‑dimensional structures de novo, without any reliance on homology templates or fragment libraries. The authors start from the observation that residues that are spatially close in a protein’s native fold tend to co‑evolve: mutations at one position are often compensated by correlated mutations at another position to preserve functional and structural integrity. While this concept has been known for decades, previous attempts to invert the evolutionary record into accurate structural constraints have largely failed because simple correlation measures conflate direct (physical) couplings with indirect statistical effects.
To overcome this, the authors employ a data‑constrained maximum‑entropy (MaxEnt) model, also known as Direct Coupling Analysis (DCA). In this framework, the probability distribution over sequences is modeled as a Boltzmann‑like distribution that reproduces the observed single‑site frequencies and pairwise frequencies from the MSA while assuming the least possible higher‑order interactions. The model parameters are the coupling strengths J_ij between each pair of positions i and j. By maximizing the pseudo‑likelihood (with L2 regularization) the authors efficiently infer J_ij for proteins of up to a few hundred residues, reducing computational cost from O(N^4) to O(N^2) or better.
The key insight is that large absolute values of J_ij correspond to “direct” statistical couplings, which are highly predictive of physical contacts in the folded structure. The authors term the resulting set of predicted contacts “Evolutionary Information Contacts” (EICs). For each protein family they select the top L·c (c≈0.2) pairs with the highest coupling scores, where L is the sequence length. These contacts are then used as distance restraints (≤8 Å between Cβ atoms, or Cα for glycine) in standard distance‑geometry and simulated‑annealing protocols (implemented in the CNS software suite). Secondary‑structure predictions (α‑helix, β‑sheet) derived from the same MSA are also incorporated to guide the folding process.
The method was tested on fifteen diverse protein families ranging from 50 to 260 residues, including a G‑protein‑coupled receptor (GPCR), a notoriously difficult target for template‑based modeling. For each target the authors generated thousands of candidate structures, scored them by violation of the EIC restraints and by a physics‑based energy function, and selected the lowest‑energy models. The resulting structures achieved Cα‑root‑mean‑square deviations (RMSD) of 2.7–5.1 Å over at least 75 % of the residues, a level of accuracy comparable to low‑resolution experimental models and sufficient for many functional analyses. Contact precision (the fraction of predicted contacts that are true contacts) ranged from 70 % to 90 %, and the authors identified a “critical number” of effective sequences (~2,000 non‑redundant members) beyond which prediction accuracy sharply improves.
Limitations are acknowledged. Small families (<1,000 effective sequences) provide insufficient statistical power, leading to noisy coupling estimates. Very large proteins (>300 residues) suffer from a scarcity of long‑range contacts relative to the number of degrees of freedom, making the restraint set underdetermined. Moreover, distance restraints alone cannot fully resolve side‑chain orientations or chirality, so the final models may require refinement with all‑atom force fields or machine‑learning‑based polishing.
The authors discuss several future directions. First, the rapid growth of metagenomic sequencing will dramatically increase the pool of divergent sequences, allowing even rare families to reach the critical depth needed for accurate DCA. Second, integrating deep neural networks that predict inter‑residue distances directly from MSAs (e.g., ResNet‑based approaches) with DCA could combine the strengths of physics‑based couplings and data‑driven patterns. Third, the pipeline can be extended to functional annotation: predicted structures can guide ligand‑binding site identification, protein‑protein interaction mapping, and the interpretation of disease‑associated missense variants.
In summary, this work demonstrates that the statistical couplings extracted from large, diverse MSAs contain sufficient information to reconstruct protein folds with near‑experimental accuracy, opening the door to a systematic, sequence‑driven structural genomics effort that could map the three‑dimensional landscape of the millions of proteins currently lacking experimental structures.
Comments & Academic Discussion
Loading comments...
Leave a Comment