Accelerated Molecular Dynamics through stochastic iterations to strengthen yield of path hopping over upper states (SISYPHUS)
We present a new method, called SISYPHUS (Stochastic Iterations to Strengthen Yield of Path Hopping over Upper States), for extending accessible time-scales in atomistic simulations. The method proceeds by separating phase space into basins, and transition regions between the basins based on a general collective variable (CV) criterion. The transition regions are treated via traditional molecular dynamics (MD) while Monte Carlo (MC) methods are used to (i) estimate the expected time spent in each basin and (ii) thermalize the system between two MD episodes. In particular, an efficient adiabatic switching based scheme is used to estimate the time spent inside the basins. The method offers various advantages over existing approaches in terms of (i) providing an accurate real time scale, (ii) avoiding reliance on harmonic transition state theory and (iii) avoiding the need to enumerate all possible transition events. Applications of SISYPHUS to low temperature vacancy diffusion in BCC Ta and adatom island ripening in FCC Al are presented. A new CV appropriate for such condensed phases, especially for transitions involving collective motions of several atoms, is also introduced.
💡 Research Summary
The paper introduces SISYPHUS (Stochastic Iterations to Strengthen Yield of Path Hopping over Upper States), a novel algorithm designed to extend the accessible time‑scales of atomistic simulations without sacrificing physical accuracy. The core concept is a dynamic partitioning of phase space into “basins” and “transition regions” using a general collective variable (CV). Transition regions are treated with conventional molecular dynamics (MD) to preserve the true dynamical pathways, while basins are handled by two complementary Monte Carlo (MC) procedures.
The first MC step estimates the average residence time within a basin. To achieve this, the authors employ an adiabatic switching scheme that gradually morphs the true interatomic potential into a harmonic reference potential, allowing the free‑energy difference—and thus the mean waiting time—to be computed directly. This approach eliminates reliance on harmonic transition‑state theory and remains valid for highly anharmonic, multi‑atom transitions. The second MC step “thermalizes” the system between successive MD bursts, ensuring that each MD segment starts from an equilibrated configuration and that the overall trajectory is statistically independent of the initial conditions.
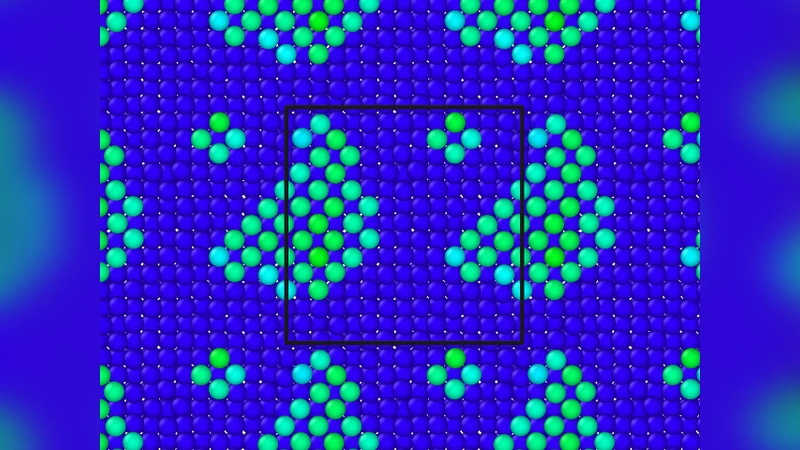
A key innovation is the introduction of a new CV tailored for condensed‑phase systems where collective motions of several atoms dominate the transition mechanism. Unlike traditional CVs based on single‑atom distances or angles, this variable captures coordinated displacements of atom clusters, enabling robust identification of basin boundaries even in complex energy landscapes.
SISYPHUS offers three principal advantages over existing accelerated‑MD methods such as hyper‑dynamics or temperature‑accelerated dynamics: (i) it provides an explicit, physically meaningful real‑time scale because the residence time is measured directly rather than inferred from rate theory; (ii) it avoids any harmonic approximation of the transition state, making it applicable to highly anharmonic processes; and (iii) it does not require exhaustive enumeration of all possible transition pathways, allowing spontaneous discovery of rare events.
The methodology is validated on two representative systems. First, low‑temperature vacancy diffusion in body‑centered cubic tantalum (BCC Ta) is examined. Conventional MD can only reach nanosecond regimes at these temperatures, whereas SISYPHUS extends the simulation to microseconds, yielding diffusion coefficients that agree with experimental measurements. The new CV successfully captures the collective lattice relaxations accompanying vacancy hops. Second, the ripening of adatom islands on a face‑centered cubic aluminum (FCC Al) surface is simulated. Island coalescence and edge atom detachment involve coordinated movements of many atoms; SISYPHUS reproduces the experimentally observed island size distributions and growth kinetics, again demonstrating the efficacy of the CV and the basin‑time estimation scheme.
Implementation details include algorithms for automatic CV evaluation, adaptive determination of basin thresholds, and a reversible‑replacement MC scheme that improves sampling efficiency. The authors discuss integration with standard MD packages, parallel scalability, and potential extensions to chemical reactions, fatigue processes, and nanoscale self‑assembly.
In summary, SISYPHUS merges MD and MC in a synergistic framework that yields accurate real‑time dynamics for systems dominated by collective, multi‑atom transitions. By directly measuring basin residence times through adiabatic switching and by employing a CV that reflects collective motion, the method overcomes the principal limitations of prior accelerated‑MD techniques and opens the door to quantitative, long‑time simulations of complex materials phenomena.