Exploring the genetic patterns of complex diseases via the integrative genome-wide approach
Motivation: Genome-wide association studies (GWASs), which assay more than a million single nucleotide polymorphisms (SNPs) in thousands of individuals, have been widely used to identify genetic risk variants for complex diseases. However, most of th…
Authors: Ben Teng, Can Yang, Jiming Liu
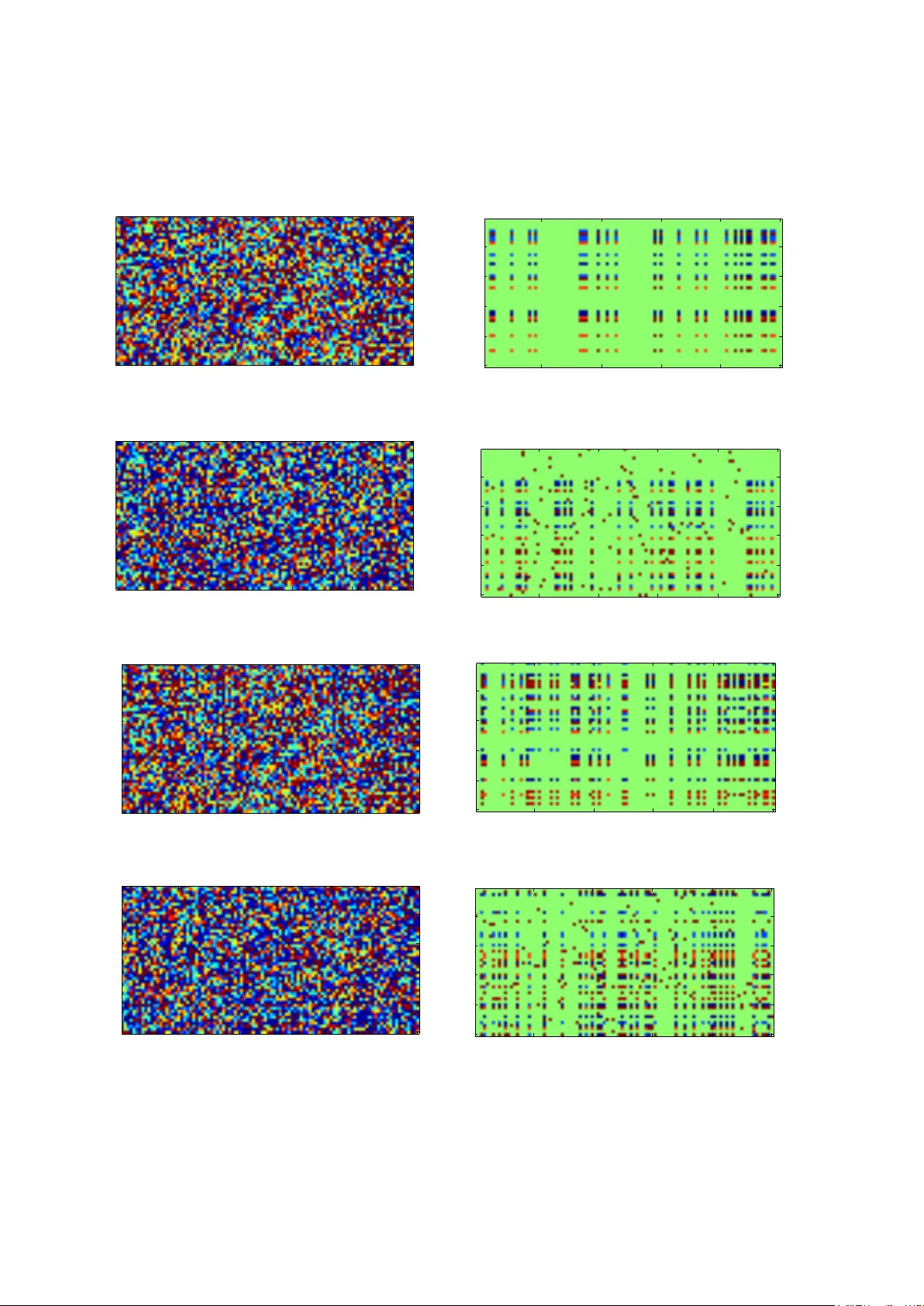
Abstract Motiv ation: Genome-wide association studies (GW ASs), which assay more than a million single nu- cleotide polymorphisms (SNPs) in thousands of individuals, hav e been widely used to identify genetic risk variants for complex diseases. Howe ver , most of the variants that have been identified contribute relativ ely small increments of risk and only explain a small portion of the genetic v ariation in complex diseases. This is the so-called missing heritability problem. Evidence has indicated that man y complex diseases are genet- ically related, meaning these diseases share common genetic risk v ariants. Therefore, exploring the genetic correlations across multiple related studies could be a promising strategy for remo ving spurious associations and identifying underlying genetic risk variants, and thereby unco vering the mystery of missing heritability in complex diseases. Results: W e present a general and robust method to identify genetic patterns from multiple large-scale genomic datasets. W e treat the summary statistics as a matrix and demonstrate that genetic patterns will form a low-rank matrix plus a sparse component. Hence, we formulate the problem as a matrix recover - ing problem, where we aim to discover risk variants shared by multiple diseases/traits and those for each individual disease/trait. W e propose a conv ex formulation for matrix recovery and an efficient algorithm to solve the problem. W e demonstrate the advantages of our method using both synthesized datasets and real datasets. The experimental results show that our method can successfully reconstruct both the shared and the individual genetic patterns from summary statistics and achieve better performance compared with alternativ e methods under a wide range of scenarios. A vailability: The MA TLAB code is av ailable at: http://www.comp.hkbu.edu.hk/ ˜ xwan/low_ rank.zip . 1 Intr oduction Many common human diseases, such as type-1 and type-2 diabetes, depression, schizophrenia, and prostate cancer , are influenced by se veral genetic and en vironmental factors. Scientists and public health officials have great interests to find genetic patterns associated with compl ex diseases, not only to adv ance our understanding of multi-gene disorders, but also to provide more insights into complex diseases. Disease association studies hav e provided substantial evidence for supporting that complex diseases originate in disorders of multiple genes [1, 2]. Nev ertheless, until recently the full-coverage identification of the genetic variants contributing to complex diseases has been staggering and dif ficult. After the completion of the Human Genome Project [3, 4] and the initiation of the International HapMap Project [5], interest has focused on genome-wide association studies (GW ASs), in which the goal is to identify single-nucleotide polymorphisms (SNPs) that are associated with complex diseases (such as diabetes) or traits (such as human height). As of Dec. 2014, more than 15 , 000 SNPs hav e been reported to be associated with at least one disease/trait at the genome-wide significance lev el ( P -v alue ≤ 5 × 10 − 8 ) [6]. Ho wev er, most of the findings only explain a small portion of the genetic contributions to complex diseases. For example, all of the 18 SNPs identified in type 2 diabetes (T2D) only account for about 6% of the inherited risk [7]. There is still 2 a large portion of disease/trait heritability that remains unexplained. This is the so-called missing heritability problem [7, 8], which is often used to denote the gap between the expected heritability of many common diseases, as estimated by family and twin studies, and the overall additiv e heritability obtained by accumulating the ef fects of all of the SNPs that have been found to be significantly associated with these conditions. A recent study [9] has suggested that most of the heritability is not missing but can be explained by the ef fects of many genetic variants, with each variant probably contributing a weak effect. Ho wever , finding v ariants with small effects is very challenging in computation because the traditional single-locus based test cannot identify such variants and the number of groups of multiple variants to be in vestigated in GW AS is astronomical. In addition, in the high-dimensional and low-sample size settings of GW AS, many irrelev ant v ariants tend to have high sample correlations due to randomness, which makes GW AS prone to false scientific discov eries. T o solv e the missing heritability problem, the large sample size is required, but such a requirement is usually beyond the capacity of a single GW AS, as the sample recruitment is expensi ve and time consuming. Evidence has indicated that many complex diseases are genetically related [10, 11, 12, 13], meaning that these diseases share common genetic risk v ariants. This suggests that an integrativ e analysis of related genomic data could be a promising strategy for removing spurious associations and identifying risk genetic variants with small effects, and thus finding the missing heritability of complex diseases. As high-throughput data acquisition becomes popular in biomedical research, new computational methods for large-scale data analysis become more and more important. When analyzing genomic data from multiple related studies, the ideal scenario is for the individual-le vel data to be av ailable for all of the included studies, but this may be difficult to achie ve due to restrictions on sharing individual-le vel data. In fact, summary data (mostly P -values or z -scores) are more frequently shared. T o identify significant SNPs shared by all of the included studies, the commonly used statistical approach is to combine P -values using Fisher’ s method [14]. [15] generalized Fisher’ s method to include weights when combining P -values. [16] suggested using the in verse normal transformation and Mosteller and [17] further generalized Stouffer’ s method by including weight when combining z -scores. There are two issues in such traditional statistical approaches. First, one small P -value can ov erwhelm many large P -values and dominate the test statistic. In a high-dimensional and low-sample size settings, many irrelev ant variants tend to hav e high significance due to randomness, which may cause wrong statistical inferences. Second, the information about genetic correlations between SNPs in the original data is completely lost after combining P -values. This information is necessary for understanding the genetic architecture of complex diseases because common complex diseases are associated with multiple genetic v ariants. T o identify shared genetic structures across multiple related studies, one feasible approach is to conduct a biclustering analysis on a matrix of summary statistics, in which the ro ws represent studies and the columns represent genetic variants, to simultaneously group studies and genetic variants. Many biclustering methods hav e been proposed and some comprehensive revie ws of biclustering methods can be found in [18], [19], and [20]. Howe ver , the traditional biclustering methods do not perform well on genomic data because genomic data 3 is high dimensional and its most genetic v ariants are irrelev ant. T o obtain sparse and interpretable biclusters, a nov el statistical approach, sparseBC , is recently proposed, which adopts an l 1 penalty to the means of the biclusters [21]. A big drawback of sparseBC is that it does not allow for ov erlapping biclusters, which limits its application in genomic data analysis because the shared genetic patterns in GW ASs may be very complex. Furthermore, in genomic data, besides the shared genetic structure, each disease/trait owns some distinct genetic v ariants. The typical biclustering model may treat them as noises and discard them. In this paper, we introduce a new method to identify genetic patterns in high dimensional genomic data. Our method possesses several advantages over existing works. First, our method admits a single model to detect both shared and individual genetic patterns among multiple studies. Second, our method employs two tuning parameters that control the size of the shared genetic pattern and the numbers of individual signals. The choices of these parameters ha ve the solid theoretical support. Third, our method produces the unique global minimizer to a con ve x problem, which means that the solution is always stable. T o demonstrate the performance of our proposed method, we conduct comparison experiments using both synthesized datasets and real datasets. Simulation results show that the proposed method outperforms existing methods in many settings. A lar ge dataset containing 32 GW ASs is also analyzed to demonstrate the advantage of our method. Specifically , we propose the con vex formulation, the algorithm, and the parameter selection in Section 2. Simulation studies and real data analysis are presented in Section 3. W e conclude the paper with some discussions in Section 4. 2 Methods 2.1 F ormulation Mathematically , the summary statistics from multiple related studies can be expressed as a matrix D ∈ R n × p , where each entry d ij is a z -score (if only P -values are av ailable, we can transform them into z -scores), and n and p are the numbers of studies and SNPs, respecti vely . Our goal is to (1) detect shared genetic patterns across studies, which can be represented as sparse biclusters in this matrix and (2) detect indi vidual genetic v ariants for each study , which we assume are randomly distributed and sparse. Since the sparsity of biclusters in a matrix indicates a low-rank property (please see e xamples in simulation studies), the problem of identifying these two types of genetic patterns can be treated as a problem of recovering a low-rank component X and a sparse component E from the input data D . Our proposed approach is based on the assumed sparsity of genetic patterns because in large-scale genomic data, most genetic v ariants are irrelev ant. W e propose to use the follo wing decomposition model to detect genetic patterns from noisy input: D = X + E + , (1) where X is a low-rank component, E is a sparse component, and is a noise component. In GW AS data analysis, the lo w-rank component corresponds to the causal SNPs that are shared by se veral diseases/traits. The 4 sparse component corresponds to the causal SNPs that af fect one specific disease/trait. The noise component corresponds to the measurement error , which is often modeled by i.i.d. Gaussian distribution with a zero mean. Naturally , to achiev e the decomposition, the following minimization problem is considered: min X , E , 1 2 k k 2 F + α rank ( X ) + β k E k 0 s.t. D = X + E + , (2) where k k F = q P i,j 2 ij is the Frobenious norm and k E k 0 is the ` 0 -norm that counts the number of nonzero v alues in E . The solution to Eq.(2) will give a penalized maximum likelihood estimate with respect to the v ariables X , E , . Ho wever , the proposed model in Eq.(2) is intractable and NP-hard. Thus, in order to effecti vely recov er X and E , we use the con ve x relaxation to replace the rank ( · ) by the nuclear norm and the ` 0 -norm by the ` 1 -norm. Here, the nuclear norm is defined as k X k ∗ = P r i =1 σ i , where σ 1 , · · · , σ r are the singular v alues of X . It is the tightest conv ex surrogate to the rank operator [22] and has been widely used for low-rank matrix recov ery [23]. The ` 1 -norm is defined as k X k 1 = P i,j | X ij | . The ` 1 relaxation has proven to be a powerful technique for sparse signal recov ery [24]. Finally , instead of directly solving Eq.(2), we solve the follo wing problem, F ( X , E ) = min X , E 1 2 k D − X − E k 2 F + α k X k ∗ + β k E k 1 . (3) It is easy to prove that Eq.(3) is a con vex problem and therefore, the global optimal solution is unique. W e will introduce the algorithm to solve this optimization problem in the ne xt subsection. 2.2 Algorithm The optimization problem of Eq.(3) can be solv ed by alternativ ely solving the following tw o sub-problems until con ver gence: ˆ X ← arg min X F ( X , ˆ E ) (4) ˆ E ← arg min E F ( ˆ X , E ) . (5) The theoretical proof for the con ver gence can be found in [25]. The problem in Eq.(4) can be reduced to min X 1 2 k D − ˆ E − X k 2 F + α k X k ∗ , (6) which becomes a nuclear-norm regularized least-squares problem and has the following closed-form solution [26], ˆ X = D α D − ˆ E , (7) 5 where D λ refers to the singular v alue thresholding (SVT) D λ ( M ) = r X i =1 ( σ i − λ ) + u i v T i . (8) Here, ( x ) + = max( x, 0) . { u i } , { v i } , and { σ i } are the left singular vectors, the right singular vectors, and the singular v alues of M , respectively . The problem in Eq.(5) can be re written as min E 1 2 k D − ˆ X − E k 2 F + β k E k 1 . (9) It admits a closed-form solution ˆ E = S β D − ˆ X , (10) where S β ( M ) ij = sign ( M ij )( M ij − β ) + refers to the elementwise soft-thresholding operator [25]. Overall, the algorithm to optimize the proposed model in Eq.(3) is summarized in Algorithm 1. It will giv e a global optimal solution independent of initialization. Algorithm 1 The algorithm to solve Eq.(3). 1. Input: D 2. Initialize all v ariables to be zero. 3. r epeat 4. Update X by solving Eq.(6) via singular v alue thresholding. 5. Update E by solving Eq.(9) via soft thresholding. 6. until con ver gence 7. Output: ˆ X and ˆ E 2.3 Parameter selection There are two parameters in our model, which can be estimated properly via the analysis of the size of the input matrix ( n, p ) and the standard variation of the noise σ [23, 27]. The relativ e weight λ = β /α balances the two terms in α k X k ∗ + β k E k 1 and consequently controls the rank of X and the sparsity of E . [23] has proved that λ = 1 / √ m gives a large probability of recov ering X and E under their assumed conditions and stated that this v alue can be adjusted slightly to obtain the best results in specific applications. Here, m is the larger dimension of the input matrix. In our problem, m = p , i.e. the number of SNPs. Howe ver , on real datasets, the shared SNPs rarely form a perfectly low-rank matrix, and we use β = 2 α/ √ p to keep suf ficient v ariations in X . The parameter α serves as a threshold in the SVT step in Eq.(8). It should be large enough to threshold out the noise but not too large to over -shrink the signal [27]. A proper value is α = ( √ n + √ p ) σ , which is the 6 expected ` 2 -norm of a n × p random matrix with entries sampled from N (0 , σ 2 ) . As SNPs are sparse in the data, we can estimate σ from the data by the median-absolute-de viation estimator [28] ˆ σ = 1 . 48 median {| D − median ( D ) |} . (11) 3 RESUL TS 3.1 Simulation studies W e first compare the performance of our method under four simulation studies, with three existing bi- clustering methods: sparseBC (sparse biclustering) [21], LAS [29] and SSVD [30]. Since biclustering methods search for sample-v ariable associations in the form of distinguished submatrices of the data matrix, we consider the entry ( i, j ) that belongs to one of the resulting biclusters which meet a predefined criterion as the reported association. Specially , for sparse biclustering method, we use the parameters that hav e been mentioned in [21], and the entries in the clusters which satisfy a preselected cutoff are recognized as the final result. For LAS, we use the default settings. For SSVD that uses a variant of singular v alue decomposition to find biclusters, we try different setting of parameters and report the best one as its result. LAS and SSVD can detect ov erlapping biclusters but sometimes they report the entire matrix as one bicluster . Thus, for both LAS and SSVD, the biclusters that contain the entire matrix are discarded. For our method, the parameters are selected as stated in Section 2. Then we use a threshold T to determine whether the entries ( i, j ) of matrix is reported as the result or not by comparing the v alue of X ( i, j ) and E ( i, j ) with T . W e e valuate each method in the term of F 1 -score, which can be calculated as follo wing: pr ecision = tp tp + f p , (12) r ecal l = tp tp + f n , (13) F 1-score = 2 ∗ pr ecision ∗ r ecal l pr ecision + r ecall , (14) where tp and f p denote the number of true positiv es and false positives, respectiv ely , and f n denotes the number of false ne gativ es. 3.1.1 Simulation settings W e adopt four patterns (each in one simulation study) illustrated in Figure 1 to generate synthetic data. • Pattern 1 adopts a case from [30], which generated a rank-1 true signal matrix. Let M = d u 1 v T 1 be a 100 × 50 matrix with d = 50 , ˆ v 1 = [10 , 9 , 8 , 7 , 6 , 5 , 4 , 3 , r (2 , 17) , r (0 , 75)] , ˆ u 1 = [10 , − 10 , 8 , − 8 , 5 , − 5 , r ( − 3 , 5) , r (0 , 34)] T , u 1 = ˆ u 1 / k ˆ u 1 k 2 , and v 1 = ˆ v 1 / k ˆ v 1 k 2 , where r ( a, b ) denotes a vector of length b with all entries equal a . This case simulates the shared causal SNPs among sev eral studies. 7 Pattern 1 20 40 60 80 100 10 20 30 40 50 Pattern 2 20 40 60 80 100 10 20 30 40 50 Pattern 3 20 40 60 80 100 10 20 30 40 50 Pattern 4 20 40 60 80 100 10 20 30 40 50 Figure 1: Four scenarios in our simulation study . Pattern 1 contains a rank-1 component representing one bicluster . Pattern 2 adds some sparse signals in Pattern 1. Pattern 3 contains a rank-2 component representing two o verlapped biclusters. Pattern 4 contains sparse signal in addition to o verlapped biclusters. 8 • Pattern 2 extends Pattern 1 by adding some sparse signals. That is, we generate a sparse component E , whose entries are independently distributed, each taking on value 0 with probability 1 − p s , and value 6 with probability p s = 0 . 01 . • Pattern 3 adopts the case from [21], which generated two overlapping biclusters. Let M = d ( u 1 v T 1 + u 2 v T 2 ) be a 100 × 50 matrix with d = 50 , u 1 and v 1 as defined in simulation 1, ˆ u 2 = [ r (0 , 13) , 10 , 9 , 8 , 7 , 6 , 5 , 4 , 3 , r (2 , 17) , r (0 , 62)] , ˆ v 2 = [ r (0 , 9) , 10 , − 9 , 8 , − 7 , 6 , − 5 , r (4 , 5) , r ( − 3 , 5) , r (0 , 25)] T , u 2 = ˆ u 2 / k ˆ u 2 k 2 , and v 2 = ˆ v 2 / k ˆ v 2 k 2 . • Pattern 4 e xtends Pattern 3 by adding some sparse signals follo wing the same way as Pattern 2. 3.1.2 Data generation Gi ven a specific pattern mentioned abov e, we first generate the data matrix. T o simulate the real situation, we randomly shuf fle the ro ws and the columns. Next, we add Gaussian noise ∼ N (0 , 1) to each item. Figure 2 illustrates the groundtruth data and the generated data. For each generated data matrix, we also compute the signal to noise ratio (SNR). T o illustrate ho w the methods perform for the data with dif ferent SNRs, we further scale do wn the ground true signal by dividing the original v alues by 1.2 and 1.5, respectiv ely . 3.1.3 Simulation results The results of four simulation studies are sho wn in Figure 3. W e use ‘lo w-rank’ to represent our method as our model is to find biclusters via a lo w-rank approximation. The details of the simulation results can be found in the supplementary materials. In general, our proposed method achiev es comparable performance in the first and third simulation studies and performs better than other three methods in the second and fourth simulation studies. This is because the classical biclustering methods suffer from sev eral limitations, such as missing some entries for ov erlapped biclusters and the inability to identify the disease/trait-specific entries. Figure 4 sho ws one result in the fourth pattern. Our proposed method can successfully recov er a low-rank component and a sparse component from raw data. In the first simulation, the F 1 -scores of sparse biclustering method and SSVD method almost get to 1. The reason why our method performs worse is that we use the default parameters which are not best fit for this simulation set-up. When adjusting the parameters, our method can also get a high F 1 -score. Furthermore, we can observe from Figure 3 that our method always perform equally well in terms of both precision and recall while the other three methods often fav or precision against recall. In the large-scale data analysis, the conservati ve method with high precision and low recall may not be suitable for new discoveries because most signals are irrelev ant. For such situations, our method has a clear advantage ov er competitors. 3.2 Real application W e applied our method to analyze 32 independent diseases/traits, including 9 20 40 60 80 100 10 20 30 40 50 Simulation 4 (Pattern 4, SNR = 2.9) 20 40 60 80 100 10 20 30 40 50 20 40 60 80 100 10 20 30 40 50 Simulation 3 (Pattern 3, SNR = 2.6) 20 40 60 80 100 10 20 30 40 50 20 40 60 80 100 10 20 30 40 50 Simulation 2 (Pattern 2, SNR = 3.3) 20 40 60 80 100 10 20 30 40 50 20 40 60 80 100 10 20 30 40 50 Simulation 1 (Pattern 1, SNR = 2.5) 20 40 60 80 100 10 20 30 40 50 Figure 2: Illustrations of four simulations. For each simulation, the generated matrix with noises is sho wn in the left panel and the groundtruth matrix is shown in the right panel. In the groundtruth matrix, the red entries indicate the true signals. 10 Precision Recall F1−score 0 0.2 0.4 0.6 0.8 1 Pattern 1 (SNR = 2.5) Precision Recall F1−score 0 0.2 0.4 0.6 0.8 1 Pattern 1 (SNR = 2.1) Precision Recall F1−score 0 0.2 0.4 0.6 0.8 1 Pattern 1 (SNR = 1.7) Precision Recall F1−score 0 0.2 0.4 0.6 0.8 1 Pattern 2 (SNR = 3.3) Precision Recall F1−score 0 0.2 0.4 0.6 0.8 1 Pattern 2 (SNR = 2.8) Precision Recall F1−score 0 0.2 0.4 0.6 0.8 1 Pattern 2 (SNR = 2.2) Precision Recall F1−score 0 0.2 0.4 0.6 0.8 1 Pattern 3 (SNR = 2.6) Precision Recall F1−score 0 0.2 0.4 0.6 0.8 1 Pattern 3 (SNR = 2.2) Precision Recall F1−score 0 0.2 0.4 0.6 0.8 1 Pattern 3 (SNR = 1.8) Precision Recall F1−score 0 0.2 0.4 0.6 0.8 1 Pattern 4 (SNR = 2.9) Precision Recall F1−score 0 0.2 0.4 0.6 0.8 1 Pattern 4 (SNR = 2.4) Precision Recall F1−score 0 0.2 0.4 0.6 0.8 1 Pattern 4 (SNR=1.9) Sparsebc SSVD LAS Low−rank Figure 3: Comparison results of dif ferent methods in four simulation studies, each using one pre-defined pat- tern. 11 Input matrix 20 40 60 80 100 10 20 30 40 50 Groundtruth matrix 20 40 60 80 100 10 20 30 40 50 Recovered low−rank component 20 40 60 80 100 10 20 30 40 50 Recovered sparse component 20 40 60 80 100 10 20 30 40 50 Figure 4: An illustration of the simulation result. The low-rank component and the sparse component are recov ered by our method. 12 • 3 anthropometrics related data. • 9 pyschiatry related data. • 8 CAD data. • 2 social science studies • 2 glycaemic traits • 7 inflammatory bo wel disease data. • systemic lupus erythematosus • parkinson The details of the data sets including the references and the web link for do wnloading the data can be found in the supplementary materials. Since each study reports different SNPs, we take the SNPs that are reported by at least 28 diseases/traits and obtain their P -v alues and impute the missing ones. Finally , we get a P -value matrix P ∈ R 466423 × 32 for these 32 diseases/traits. Next, we con vert the P -value matrix to the z -score matrix Z ∈ R 466423 × 32 . W e analyze this data set using our method on a desktop PC with 2.40GHz CPU and 4GB RAM. The running time of our method on 32 GW ASs data sets is only 152.1s. The three alterativ e methods in vestigated in this w ork cannot be applied due to the large size of the data. The experiment results are gi ven in Figure 5. The shared causal SNPs are presented in the lo w-rank com- ponent and indi vidual-specific SNPs are shown in the sparse component. W e take the first three right singular vectors of the recov ered lo w-rank matrix and use them as the coordinate of each study in Figure 6. From Figure 6, it is clear to see that 3 clusters are recov ered from 32 diseases/traits: • 2 social science studies (edu years and college); • diastolic blood pressure and systolic blood pressure (DBP and SBP); • total cholesterol and lo w density lipoprotein (TC and LDL). The diseases/traits in each cluster are highly relev ant with each other . W e compare the identified causal SNPs by our method on 32 GW AS data with some previous findings. For 3 pairs of diseases/traits that are clustered together, we mainly in vestigate the shared SNPs that are identified by our method. For two social science related data, our method has detected SNP rs3789044 , SNP rs12046747 , and SNP rs12853561 , which are mapped to genes LRRN2 and STK24 , respectively . These were reported in the original article [31] because they hav e significant P -v alues (the details are provided in the supplementary materials). Ho wev er, besides those SNPs with significant P -values, our method has also identified some locus with moderate P -v alues. SNP rs2532269 , whose original P -values are 1 . 01 × 10 − 4 in edu years data and 1 . 11 × 10 − 4 in college data, is detected as a causal SNP by our method. This SNP was previously reported ( P -value = 2 × 10 − 11 ) [32] and 13 Input matrix Diseases/traits SNPs Low rank component Diseases/traits SNPs Figure 5: The experiment results on 32 GW ASs. The low-rank component (middle panel) and the sparse component (right panel) are recov ered by our method. mapped to the gene KIAA1267 . This gene is highly connected with Koolen-De Vries syndrome. K oolen-De Vries syndrome is characterized by moderate to se vere intellectual disability , hypotonia, friendly demeanor , and highly distincti ve facial features, including tall, broad forehead, long face, upslanting palpebral fissures, epicanthal folds, tubular nose with b ulbous nasal tip, and large ears [33]. For diastolic blood pressure and systolic blood pressure, the identified SNPs in our experiment are also connected with some previously published genes, such as ULK4, FGF5 and C10orf107 [34]. Similarly , some additional locus are identified by the low-rank component. SNP rs4986172 (original P -values in SBP data and DBP data are 3 . 09 × 10 − 5 and 0 . 0172 , respectively), located in the gene ACBD4 , is detected by the low-rank component. This gene has been associated with high blood pressure in [35]. T o illustrate the power of our method in identifying the causal SNPs that do not shared by sev eral dis- eases/traits, we take the result of bipolar disorder as an example. The SNPs in the result of bipolar disorder can be matched to ANK3, CACN A1C, SYNE1 and PBRM1 , which have been confirmed to be associated with bipolar disorder [36]. The detailed results of other diseases/traits can be found in the supplementary materials. Clearly , the experiment results sho w that not only can our method recognize SNPs with small P -values, but also detect those SNPs with moderate or weak P -v alues. 14 −0.8 −0.6 −0.4 −0.2 0 −1 −0.5 0 0.5 −0.2 0 0.2 0.4 0.6 0.8 DBP and SBP edu_years and College LDL and TC Figure 6: The geometric relationships of all studies using the coordinates deriv ed from the first three right singular vectors of the reco vered lo w-rank matrix. 4 Discussion Finding weak-effect variants to explain the missing heritability of complex diseases is a challenging task and bottlenecked by the av ailable sample size of GW AS. Based on the fact that related diseases/traits tend to co-occur , discov ering shared genetic components among related studies becomes a popular way to address this issue. In the last fe w years, hundreds of GW ASs ha ve been carried out. Therefore, it is timely to systematically in vestigate GW AS data sets to find those shared patterns for comprehensi ve understanding of the genetic archi- tecture of complex diseases/traits. In this work, we present a no vel method for exploring the genetic patterns of complex diseases. W e assume that causal SNPs can be di vided into two categories: SNPs shared by multiple diseases/traits and SNPs for individual disease/trait. Thus, by modeling the problem as recovering a low-rank component and a sparse component from a noise matrix, we formulate it as a conv ex optimization problem. T o demonstrate the performance of our proposed method, we conduct sev eral simulation studies under different settings. Simulation results show that the proposed method outperforms three alternati ve methods in man y set- tings. In the real data studies, we collect 32 large-scale GW AS data sets. W e hav e successiv ely analyzed these data sets via our proposed method and discov ered some interesting shared genetic patterns. Many identified v ariants hav e been confirmed by other works. T o conclude, our proposed method not only possesses a better po wer than related methods but also provides easily interpretable results for better understanding shared genetic architectures of complex diseases/trais. 15 In this work, we mainly focus on the analysis of summary statistics. W ith the dev elopment of new tech- nology , more and more supplementary information, such as functional annotation data, structural data, and biochemical data, can be quickly obtained. In the future work, we will integrate these information in our method to increase the statistical po wer . Acknowledgments This w ork was supported by Georgia State Univ ersity Deep Grant, Hong Kong Baptist Univ ersity Strategic De velopment Fund, Hong K ong Baptist Univ ersity grant, and Hong Kong Research grant HKB U12202114. 16 Supplementary Document f or “Exploring the genetic patterns of complex diseases via the integrative genome-wide approach” List • T able S1: 32 GW ASs data. • T able S2: 32 GW ASs data. • T able S3: Results of four methods when simulations are generated from pattern 1 with different SNRs. • T able S4: Results of four methods when simulations are generated from pattern 2 with different SNRs. • T able S5: Results of four methods when simulations are generated from pattern 3 with different SNRs. • T able S6: Results of four methods when simulations are generated from pattern 4 with different SNRs. 17 Data descriptions W e applied our method to analyze 32 independent diseases/traits, including • 3 anthropometrics related data: body mass index [37], height [38], w aist-hip ratio adjusted for BMI [39]. • 9 pyschiatry related data: fiv e PGC data [13] (attention-deficit/hyperactivity disorder , autism spectrum disorder , bipolar disorder , major depressiv e disorder , schizophrenia) and four T A G data [40] (T agCPD, T agEVRSMK, T agFORMER, T agLOGONSET). • 8 CAD data: total cholesterol [41], low density lipoprotein [41], triglycerides [41], high density lipopro- tein [41], type 2 diabetes [2], coronary artery disease [42], diastolic blood pressure [34], systolic blood pressure [34]. • 2 social science related data [31]: edu years, college. • 2 glycaemic traits [43]: fasting glucose, fasting insulin. • 7 inflammatory bo wel disease data: crohn’ s disease [44], multiple sclerosis [45], psoriasis [46], rheuma- toid arthritis [47], type 1 diabetes [48], ulcerati ve colitis [49]. • systemic lupus erythematosus [50]. • parkinson [51]. 18 T able 1: 32 GW ASs data. Name # of SNPs Link body mass index [37] 2471516 http://www.broadinstitute.org/collaboration/ giant/index.php/ height [38] 2469635 http://www.broadinstitute.org/collaboration/ giant/index.php/ crohn’ s disease [44] 953241 http://www.ibdgenetics.org/downloads.html fasting glucose [43] 2628879 http://www.magicinvestigators.org/downloads/ total cholesterol [41] 2693413 http://www.sph.umich.edu/csg/abecasis/public/ lipids2010/ lo w density lipopro- tein [41] 2692564 http://www.sph.umich.edu/csg/abecasis/public/ lipids2010/ triglycerides [41] 2692560 [41] http://www.sph.umich.edu/csg/abecasis/public/ lipids2010/ high density lipopro- tein [41] 2692429 http://www.sph.umich.edu/csg/abecasis/public/ lipids2010/ coronary artery dis- ease [42] 2420360 http://www.cardiogramplusc4d.org/downloads/ college [31] 2321510 http://ssgac.org/Data.php diastolic blood pres- sure [34] 2461325 http://www.ncbi.nlm.nih.gov/projects/gap/ cgi- bin/study.cgi?study\_id=phs000585.v1.p1 systolic blood pressure [34] 2461325 http://www.ncbi.nlm.nih.gov/projects/gap/ cgi- bin/study.cgi?study\_id=phs000585.v1.p1 eduyears [31] 2310087 http://ssgac.org/Data.php fasting Insulin [43] 2627848 http://www.magicinvestigators.org/downloads/ multiple sclerosis [45] 327094 http://www.ncbi.nlm.nih.gov/projects/gap/ cgi- bin/analysis.cgi?study\_id=phs000139.v1.p1\ &phv=65549\&phd=1061\&pha=2854\&pht=621\&phvf= \&phdf=\&phaf=\&phtf=\&dssp=1\&consent=\&temp=1 parkinson [51] 453217 http://www.ncbi.nlm.nih.gov/projects/gap/ cgi- bin/analysis.cgi\?study\_id=phs000089.v3. p2&phv=24040&phd=392&pha=2868&pht=178&phvf= &phdf=&phaf=&phtf=&dssp=1&consent=&temp=1 19 T able 2: 32 GW ASs data. Name # of SNPs Link attention- deficit/hyperacti vity disorder [13] 1219805 http://www.med.unc.edu/pgc/downloads autism spectrum disor- der [13] 1219805 http://www.med.unc.edu/pgc/downloads bipolar disorder [13] 1219805 http://www.med.unc.edu/pgc/downloads major depressi ve dis- order [13] 1219805 http://www.med.unc.edu/pgc/downloads schizophrenia [13] 1219805 http://www.med.unc.edu/pgc/downloads psoriasis [46] 440153 http://www.ncbi.nlm.nih.gov/projects/gap/ cgi- bin/analysis.cgi\?study_id=phs000019.v1. p1\&phv=20012\&phd=179\&pha=2855\&pht=63\&phvf= \&phdf=\&phaf=\&phtf=\&dssp=1\&consent=\&temp=1 rheumatoid arthritis [47] 2556271 http://www.broadinstitute.org/ftp/pub/ rheumatoid\_arthritis/Stahl\_etal\_2010NG/ type 1 diabetes [48] 503181 http://www.ncbi.nlm.nih.gov/projects/gap/ cgi- bin/analysis.cgi\?study_id=phs000180.v2.p2\ &phv=73462\&phd=1548\&pha=2862\&pht=789\&phvf= \&phdf=\&phaf=\&phtf=\&dssp=1\&consent=\&temp=1 type 2 diabetes [2] 2473441 http://diagram- consortium.org/downloads.html T agCPD [40] 2459118 http://www.med.unc.edu/pgc/downloads T agEVRSMK [40] 2455846 http://www.med.unc.edu/pgc/downloads T agFORMER [40] 2456554 http://www.med.unc.edu/pgc/downloads T agLOGONSET [40] 2457545 http://www.med.unc.edu/pgc/downloads ulcerati ve colitis [49] 1428749 http://www.ibdgenetics.org/ waist-hip ratio ad- justed for BMI [39] 2483326 http://www.broadinstitute.org/collaboration/ giant/index.php/GIANT\_consortium\_data\_files systemic lupus erythe- matosus [50] 258402 http://www.ncbi.nlm.nih.gov/projects/gap/ cgi- bin/analysis.cgi?study_id=phs000122.v1. p1&phv=66336&phd=&pha=2848&pht=629&phvf=&phdf= &phaf=&phtf=&dssp=1&consent=&temp=1 20 T able 3: Results of four methods when simulations are generated fr om pattern 1 with different SNRs. SNR = 2 . 5 SNR = 2 . 1 SNR = 1 . 7 Sparsebc SSVD LAS Low-rank Sparsebc SSVD LAS Lo w-rank Sparsebc SSVD LAS Low-rank Precision 0.95 0.95 1 0.84 0.88 0.94 1 0.75 0.71 0.92 1 0.61 Recall 0.95 0.99 0.19 0.82 0.96 0.94 0.17 0.82 0.70 0.76 0.15 0.82 F1-score 0.95 0.96 0.32 0.83 0.92 0.94 0.29 0.78 0.70 0.82 0.26 0.70 T able 4: Results of four methods when simulations are generated fr om pattern 2 with different SNRs. SNR = 3 . 3 SNR = 2 . 8 SNR = 2 . 2 Sparsebc SSVD LAS Low-rank Sparsebc SSVD LAS Lo w-rank Sparsebc SSVD LAS Low-rank Precision 0.72 0.80 0.98 0.85 0.69 0.87 0.97 0.75 0.72 0.80 1 0.69 Recall 0.68 0.80 0.27 0.85 0.57 0.72 0.22 0.86 0.46 0.63 0.18 0.74 F1-score 0.69 0.80 0.42 0.85 0.61 0.79 0.36 0.80 0.56 0.71 0.30 0.71 T able 5: Results of four methods when simulations are generated fr om pattern 3 with different SNRs. SNR = 2 . 6 SNR = 2 . 2 SNR = 1 . 8 Sparsebc SSVD LAS Low-rank Sparsebc SSVD LAS Lo w-rank Sparsebc SSVD LAS Low-rank Precision 0.99 0.74 1 0.84 0.85 0.74 1 0.75 0.87 0.78 1 0.77 Recall 0.61 0.79 0.29 0.86 0.63 0.79 0.22 0.84 0.56 0.69 0.22 0.75 F1-score 0.76 0.77 0.45 0.85 0.73 0.76 0.37 0.79 0.68 0.73 0.37 0.76 T able 6: Results of four methods when simulations are generated fr om pattern 4 with different SNRs. SNR = 2 . 9 SNR = 2 . 4 SNR = 1 . 9 Sparsebc SSVD LAS Low-rank Sparsebc SSVD LAS Lo w-rank Sparsebc SSVD LAS Low-rank Precision 0.86 0.76 0.99 0.80 0.81 0.85 1 0.78 0.75 0.86 1 0.71 Recall 0.64 0.68 0.27 0.83 0.61 0.48 0.20 0.76 0.50 0.48 0.19 0.71 F1-score 0.72 0.72 0.43 0.82 0.69 0.62 0.23 0.77 0.59 0.62 0.32 0.71 21 Refer ences [1] McClellan JM, Susser E, King MC: Schizophrenia: a common disease caused by multiple rar e alleles . The British Journal of Psychiatry 2007, 190 (3):194–199. [2] Morris AP , V oight BF , T eslovich TM, Ferreira T , Segre A V , Steinthorsdottir V , Strawbridge RJ, Khan H, Grallert H, Mahajan A, et al.: Large-scale association analysis pro vides insights into the genetic architectur e and pathophysiology of type 2 diabetes . Natur e genetics 2012, 44 (9):981. [3] V enter JC, Adams MD, Myers EW , Li PW , Mural RJ, Sutton GG, Smith HO, Y andell M, Ev ans CA, Holt RA, et al.: The sequence of the human genome . science 2001, 291 (5507):1304–1351. [4] Lander E, Linton L, Birren B, Nusbaum C, Zody M, Baldwin J, Dev on K, Dew ar K, Doyle M, FitzHugh W , et al.: Initial sequencing and analysis of the human genome . Nature 2001, 409 (6822):860–921. [5] Sachidanandam R, W eissman D, Schmidt S, Kakol J, Stein L, Marth G, Sherry S, Mullikin J, Morti- more B, W illey D, et al.: A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms . Natur e 2001, 409 (6822):928–933. [6] Hindorf f L, Junkins H, Hall P , Mehta J, Manolio T : A Catalog of Published Genome-W ide Association Studies. A vailable at: www .genome.gov/gwastudies. Accessed J anuary 22, 2015 2015. [7] Manolio T , Collins F , Cox N, Goldstein D, Hindorff L, Hunter D, McCarthy M, Ramos E, Car- don L, Chakrav arti A, et al.: Finding the missing heritability of complex diseases . Nature 2009, 461 (7265):747–753. [8] Maher B: P ersonal genomes: The case of the missing heritability . Nature 2008, 456 (7218):18. [9] Y ang J, Benyamin B, McEvoy BP , Gordon S, Henders AK, Nyholt DR, Madden P A, Heath A C, Martin NG, Montgomery GW , et al.: Common SNPs explain a large proportion of the heritability for human height . Natur e genetics 2010, 42 (7):565–569. [10] Si vakumaran S, Agakov F , Theodoratou E, Prendergast JG, Zgaga L, Manolio T , Rudan I, McKeigue P , W ilson JF , Campbell H: Abundant pleiotropy in human complex diseases and traits . The American J ournal of Human Genetics 2011, 89 (5):607–618. [11] V attikuti S, Guo J, Chow CC: Heritability and genetic correlations explained by common SNPs f or metabolic syndrome traits . PLoS genetics 2012, 8 (3):e1002637. [12] Consortium PG, et al.: Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs . Natur e genetics 2013. 22 [13] Cross-Disorder Group of the Psychiatric Genomics Consortium, et al.: Identification of risk loci with shar ed effects on five major psychiatric disorders: a genome-wide analysis . Lancet 2013, 381 (9875):1371. [14] Fisher RA: Statistical methods f or research w orkers 1934. [15] Goods I: On the weighted combination of significance tests . Journal of the Royal Statistical Society . Series B (Methodological) 1955, :264–265. [16] Stouf fer SA, Suchman EA, DeV inney LC, Star SA, W illiams Jr RM: The Am erican soldier: adjustment during army life.(Studies in social psychology in World War II, Vol. 1.). 1949. [17] Mosteller F , Bush RR, Green BF: Selected quantitative techniques . Addison-W esley 1970. [18] Madeira SC, Oli veira AL: Biclustering algorithms for biological data analysis: a survey . Computa- tional Biology and Bioinformatics, IEEE/ACM T ransactions on 2004, 1 :24–45. [19] Preli ´ c A, Bleuler S, Zimmermann P , W ille A, B ¨ uhlmann P , Gruissem W , Hennig L, Thiele L, Zitzler E: A systematic comparison and evaluation of biclustering methods f or gene expr ession data . Bioinfor - matics 2006, 22 (9):1122–1129. [20] Busygin S, Prok opyev O, Pardalos PM: Biclustering in data mining . Computers & Operations Resear ch 2008, 35 (9):2964–2987. [21] T an KM, W itten DM: Sparse biclustering of transposable data . J ournal of Computational and Graphi- cal Statistics 2013, (just-accepted). [22] Fazel M: Matrix rank minimization with applications . PhD thesis , Stanford Univ ersity 2002. [23] Cand ` es E, Li X, Ma Y , Wright J: Robust principal component analysis? Journal of the A CM 2011, 58 (3):11. [24] T ropp J A: Just relax: Con vex programming methods f or identifying sparse signals in noise . IEEE T ransactions on Information Theory 2006, 52 (3):1030–1051. [25] Boyd S: Distrib uted Optimization and Statistical Learning via the Alternating Dir ection Method of Multipliers . F oundations and T r ends R in Machine Learning 2010, 3 :1–122. [26] Cai J, Cand ` es E, Shen Z: A Singular V alue Thresholding Algorithm for Matrix Completion . SIAM J ournal on Optimization 2010, 20 :1956. [27] Zhou Z, Li X, Wright J, Candes E, Ma Y : Stable principal component pursuit . In Pr oceedings of the IEEE International Symposium on Information Theory 2010. 23 [28] Meer P , Mintz D, Rosenfeld A, Kim D: Robust regression methods for computer vision: A review . International J ournal of Computer V ision 1991, 6 :59–70. [29] Shabalin AA, W eigman VJ, Perou CM, Nobel AB: Finding large average submatrices in high dimen- sional data . The Annals of Applied Statistics 2009, :985–1012. [30] Lee M, Shen H, Huang JZ, Marron J: Biclustering via sparse singular value decomposition . Biometrics 2010, 66 (4):1087–1095. [31] Rietveld CA, Medland SE, Derringer J, Y ang J, Esko T , Martin NW , W estra HJ, Shakhbazov K, Abdel- laoui A, Agrawal A, et al.: GW AS of 126,559 individuals identifies genetic variants associated with educational attainment . Science 2013, 340 (6139):1467–1471. [32] Consortium EGGE, et al.: Common variants at 6q22 and 17q21 are associated with intracranial volume . Natur e g enetics 2012, 44 (5):539–544. [33] K oolen DA, Kramer JM, Neveling K, Nillesen WM, Moore-Barton HL, Elmslie FV , T outain A, Amiel J, Malan V , Tsai ACH, et al.: Mutations in the chromatin modifier gene KANSL1 cause the 17q21. 31 microdeletion syndr ome . Natur e genetics 2012, 44 (6):639–641. [34] International Consortium for Blood Pressure Genome-W ide Association Studies, et al.: Genetic vari- ants in novel pathways influence blood pressur e and cardiovascular disease risk . Natur e 2011, 478 (7367):103–109. [35] Ne wton-Cheh C, Johnson T , Gatev a V , T obin MD, Bochud M, Coin L, Najjar SS, Zhao JH, Heath SC, Eyheramendy S, et al.: Eight blood pressur e loci identified by genome-wide association study of 34,433 people of European ancestry . Natur e genetics 2009, 41 (6):666. [36] Chung D, Y ang C, Li C, Gelernter J, Zhao H: GP A: A Statistical Appr oach to Prioritizing GW AS Results by Integrating Pleiotropy and Annotation . PLoS genetics 2014, 10 (11):e1004787. [37] Speliotes EK, W iller CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson A U, Allen HL, Lindgren CM, Luan J, M ¨ agi R, et al.: Association analyses of 249,796 individuals r eveal 18 new loci associated with body mass index . Natur e genetics 2010, 42 (11):937–948. [38] Allen HL, Estrada K, Lettre G, Berndt SI, W eedon MN, Ri vadeneira F , W iller CJ, Jackson A U, V edantam S, Raychaudhuri S, et al.: Hundreds of variants clustered in genomic loci and biological pathways affect human height . Natur e 2010, 467 (7317):832–838. [39] Heid IM, Jackson A U, Randall JC, Winkler TW , Qi L, Steinthorsdottir V , Thorleifsson G, Zillikens MC, Speliotes EK, M ¨ agi R, et al.: Meta-analysis identifies 13 new loci associated with waist-hip ratio and re veals sexual dimorphism in the genetic basis of fat distribution . Natur e genetics 2010, 42 (11):949– 960. 24 [40] T obacco and Genetics Consortium, et al.: Genome-wide meta-analyses identify multiple loci associated with smoking beha vior . Nature genetics 2010, 42 (5):441–447. [41] T eslovich TM, Musunuru K, Smith A V , Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP , Ripatti S, Chasman DI, Willer CJ, et al.: Biological, clinical and population rele vance of 95 loci for blood lipids . Natur e 2010, 466 (7307):707–713. [42] Deloukas P , Kanoni S, Willenbor g C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein B A, et al.: Large-scale association analysis identifies new risk loci f or coronary artery disease . Natur e genetics 2013, 45 :25–33. [43] Manning AK, Hiv ert MF , Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, Rybin D, Liu CT , Bielak LF , Prokopenko I, et al.: A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance . Natur e genetics 2012, 44 (6):659– 669. [44] Jostins L, Ripke S, W eersma RK, Duerr RH, McGov ern DP , Hui KY , Lee JC, Schumm LP , Sharma Y , Anderson CA, et al.: Host-microbe interactions hav e shaped the genetic architectur e of inflammatory bowel disease . Natur e 2012, 491 (7422):119–124. [45] Fingerprinting G: Risk alleles f or multiple sclerosis identified by a genomewide study . N engl J med 2007, 357 :851–862. [46] Feng BJ, Sun LD, Soltani-Arabshahi R, Bowcock AM, Nair RP , Stuart P , Elder JT , Schrodi SJ, Begovich AB, Abecasis GR, et al.: Multiple loci within the major histocompatibility complex confer risk of psoriasis . PLoS genetics 2009, 5 (8):e1000606. [47] Stahl EA, Raychaudhuri S, Remmers EF , Xie G, Eyre S, Thomson BP , Li Y , Kurreeman F A, Zhernako va A, Hinks A, et al.: Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci . Natur e genetics 2010, 42 (6):508–514. [48] Barrett JC, Clayton DG, Concannon P , Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, et al.: Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes . Natur e genetics 2009, 41 (6):703–707. [49] Anderson CA, Boucher G, Lees CW , Franke A, D’Amato M, T aylor KD, Lee JC, Goyette P , Imielinski M, Latiano A, et al.: Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47 . Natur e genetics 2011, 43 (3):246–252. [50] Hom G, Graham RR, Modrek B, T aylor KE, Ortmann W , Garnier S, Lee A T , Chung SA, Ferreira RC, P ant PK, et al.: Association of systemic lupus erythematosus with C8orf13–BLK and ITGAM–ITGAX . New England J ournal of Medicine 2008, 358 (9):900–909. 25 [51] Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P , Scholz SW , Hernandez DG, et al.: Genome-wide association study rev eals genetic risk underlying Parkin- son’ s disease . Natur e genetics 2009, 41 (12):1308–1312. 26
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment