Feature extraction from complex networks: A case of study in genomic sequences classification
This work presents a new approach for classification of genomic sequences from measurements of complex networks and information theory. For this, it is considered the nucleotides, dinucleotides and trinucleotides of a genomic sequence. For each of them, the entropy, sum entropy and maximum entropy values are calculated.For each of them is also generated a network, in which the nodes are the nucleotides, dinucleotides or trinucleotides and its edges are estimated by observing the respective adjacency among them in the genomic sequence. In this way, it is generated three networks, for which measures of complex networks are extracted.These measures together with measures of information theory comprise a feature vector representing a genomic sequence. Thus, the feature vector is used for classification by methods such as SVM, MultiLayer Perceptron, J48, IBK, Naive Bayes and Random Forest in order to evaluate the proposed approach.It was adopted coding sequences, intergenic sequences and TSS (Transcriptional Starter Sites) as datasets, for which the better results were obtained by the Random Forest with 91.2%, followed by J48 with 89.1% and SVM with 84.8% of accuracy. These results indicate that the new approach of feature extraction has its value, reaching good levels of classification even considering only the genomic sequences, i.e., no other a priori knowledge about them is considered.
💡 Research Summary
The paper introduces a novel feature extraction framework that combines complex network analysis with information‑theoretic measures to classify genomic sequences. The authors first decompose each DNA sequence into three levels of k‑mers: nucleotides (1‑mer), dinucleotides (2‑mer), and trinucleotides (3‑mer). For each level they construct a graph where nodes correspond to the distinct k‑mers and edges represent adjacency relationships observed in the original sequence. Edge creation is governed by two parameters: word size (WS), which defines the length of the k‑mer, and step (P), which determines how many characters are skipped before the next k‑mer is read. By varying WS ∈ {1,2,3} and P ∈ {1,2,3}, six distinct undirected networks are generated (one for 1‑mer, two for 2‑mer, and three for 3‑mer).
From each network the authors extract a comprehensive set of complex‑network descriptors: average shortest‑path length, clustering coefficient, several centrality metrics (degree, closeness, betweenness, efficiency), average degree, degree statistics (mean, standard deviation, min, max), motif frequencies, and the number of communities detected via modularity‑based clustering. In parallel, they compute information‑theoretic features for the same three k‑mer levels: Shannon entropy, the sum of entropies, and the maximal entropy obtained through a constrained optimization formulation. All these values are concatenated into a high‑dimensional feature vector that uniquely represents each genomic fragment.
The methodology is evaluated on three biologically relevant datasets: (1) 1,600 human protein‑coding sequences and 1,520 non‑coding (intergenic) regions extracted from RefSeq, (2) 1,500 transcription‑start‑site (TSS) promoter sequences from the DBTSS database, and (3) a combined set used for cross‑validation. Classification is performed using six standard machine‑learning algorithms implemented in the WEKA suite: Random Forest, J48 decision tree, Support Vector Machine (with an RBF kernel), Multi‑Layer Perceptron, IBk (k‑nearest neighbours), and Naïve Bayes. Ten‑fold cross‑validation is applied, and all extracted features are fed to each classifier without prior feature selection.
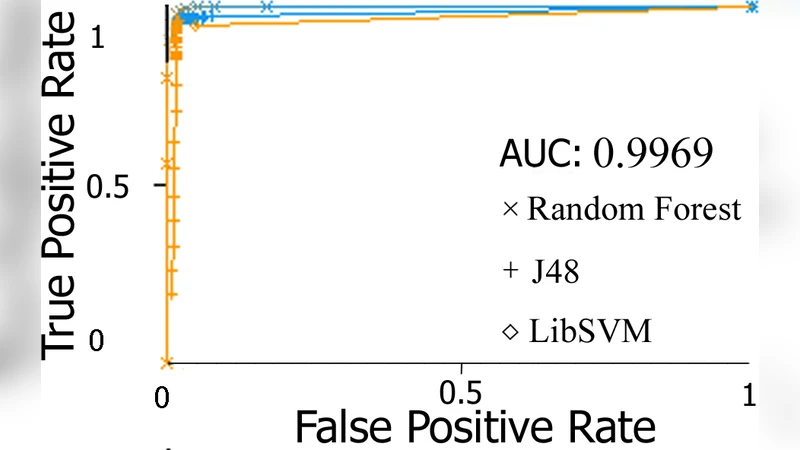
Results show that the Random Forest classifier achieves the highest overall accuracy of 91.2 %, followed closely by J48 (89.1 %) and SVM (84.8 %). The remaining algorithms attain lower but still respectable performance (MLP = 64.5 %, Naïve Bayes = 72.6 %, IBk = 79.9 %). Receiver‑Operating‑Characteristic (ROC) curves for each class (coding, intergenic, promoter) confirm the robustness of the feature set, with especially high AUC values for the intergenic class, which exhibits pronounced nucleotide repetition and thus distinctive network topology.
The authors discuss several advantages of their approach. First, it relies solely on raw sequence data, requiring no external annotations or prior biological knowledge. Second, the WS and P parameters provide a flexible mechanism to generate multiple network representations, potentially enriching the feature space and improving classification when more combinations are explored. Third, the combination of global information‑theoretic descriptors with local network‑based metrics captures complementary aspects of sequence organization, leading to more discriminative representations than either type of feature alone. Finally, the framework is readily extensible to other genomic entities such as micro‑RNAs, transposons, protein‑coding genes, and regulatory elements, suggesting broad applicability in bioinformatics and systems biology.
In summary, this study demonstrates that integrating complex‑network measures with entropy‑based statistics yields a powerful, annotation‑free feature extraction pipeline capable of high‑accuracy classification of diverse genomic regions. The work opens avenues for further methodological refinements (e.g., automated parameter optimization, dimensionality reduction, deep‑learning integration) and for applying the technique to larger, more heterogeneous genomic datasets.
Comments & Academic Discussion
Loading comments...
Leave a Comment