Host Cell Factors Necessary for Influenza A Infection: Meta-Analysis of Genome Wide Studies
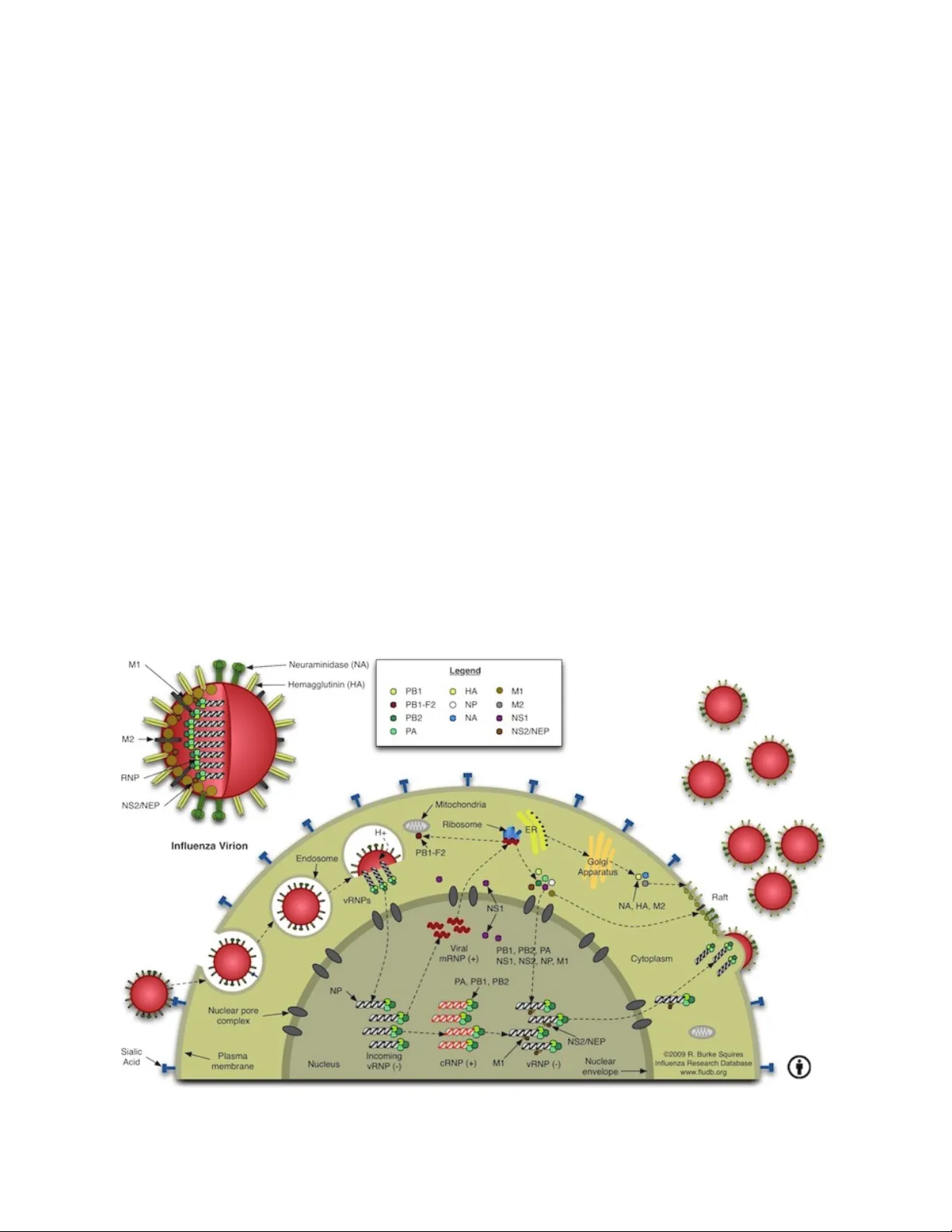
The Influenza A virus belongs to the Orthomyxoviridae family. Influenza virus infection occurs yearly in all countries of the world. It usually kills between 250,000 and 500,000 people and causes severe illness in millions more. Over the last century alone we have seen 3 global influenza pandemics. The great human and financial cost of this disease has made it the second most studied virus today, behind HIV. Recently, several genome-wide RNA interference studies have focused on identifying host molecules that participate in Influenza infection. We used nine of these studies for this meta-analysis. Even though the overlap among genes identified in multiple screens was small, network analysis indicates that similar protein complexes and biological functions of the host were present. As a result, several host gene complexes important for the Influenza virus life cycle were identified. The biological function and the relevance of each identified protein complex in the Influenza virus life cycle is further detailed in this paper.
💡 Research Summary
Influenza A virus remains a major global health threat, causing up to half a million deaths each year and prompting periodic pandemics. Over the past decade, nine independent genome‑wide RNA interference (RNAi) screens have been performed to identify host factors that facilitate influenza infection. Although the overlap of individual hit genes among these studies is modest, the authors of this meta‑analysis leveraged protein‑protein interaction (PPI) networks to uncover higher‑order commonalities. By mapping each screen’s statistically significant hits onto high‑confidence human PPI databases (STRING, HINT, BioGRID) and applying network‑centric metrics (betweenness, clustering coefficient) together with module detection algorithms (e.g., MCODE), they identified a set of densely connected hubs and functional modules that recur across the datasets.
The analysis revealed that, despite only ~30 shared genes out of >1,200 total hits, these genes cluster into several biologically coherent complexes: (1) nuclear pore and export complexes (NUP98, NUP153, NXF1/TAP) that mediate viral ribonucleoprotein (vRNP) nuclear‑cytoplasmic trafficking; (2) COPI/COPII vesicular transport machinery (ARF1, SEC23/24) essential for endosomal escape and virion assembly; (3) spliceosome components (SF3B, U2AF, PRPF8) that intersect with viral mRNA processing; (4) translation initiation factors (eIF3, eIF4F, eIF4E) required for cap‑dependent synthesis of viral proteins; and (5) ribosome biogenesis and quality‑control factors (RPL/RPS families, NOB1, DDX3). Gene Ontology and KEGG enrichment further highlighted pathways such as RNA processing, intracellular transport, cytoskeletal remodeling, and interferon signaling.
By cross‑referencing these complexes with the known stages of the influenza life cycle, the authors mapped each module to a specific functional window: the clathrin‑coated vesicle and V‑ATPase complex to viral entry and endosomal acidification; the nuclear export machinery to vRNP export; the spliceosome and transcription factors to viral mRNA maturation; and the translation apparatus to viral protein production. Importantly, several identified complexes—GPI‑anchor biosynthesis enzymes, V‑ATPase subunits, and ribosome quality‑control proteins—have not yet been explored as antiviral targets.
To prioritize candidates for follow‑up validation, the study assigned a “core dependency score” to ~50 host genes that consistently appeared in multiple networks. The authors propose secondary validation using CRISPR‑KO or siRNA knock‑down coupled with viral replication assays, and suggest that these high‑confidence hits could serve as novel drug targets or be repurposed from existing pharmacophores.
In summary, this meta‑analysis transcends the limitations of individual RNAi screens by integrating them into a systems‑level interaction map. The convergence on specific protein complexes and pathways underscores the virus’s reliance on a relatively small set of host machineries, despite the apparent diversity of individual genetic hits. These insights provide a robust framework for the development of next‑generation host‑targeted antivirals against influenza A and illustrate the power of network‑based meta‑analysis in virology research.
Comments & Academic Discussion
Loading comments...
Leave a Comment