A Gauge Field Theory of Chirally Folded Homopolymers with Applications to Folded Proteins
We combine the principle of gauge invariance with extrinsic string geometry to develop a lattice model that can be employed to theoretically describe properties of chiral, unbranched homopolymers. We find that in its low temperature phase the model is in the same universality class with proteins that are deposited in the Protein Data Bank, in the sense of the compactness index. We apply the model to analyze various statistical aspects of folded proteins. Curiously we find that it can produce results that are a very good good match to the data in the Protein Data Bank.
💡 Research Summary
The paper introduces a novel lattice model for chiral, unbranched homopolymers by merging the principle of gauge invariance with the extrinsic geometry of strings. Starting from a continuous description of a polymer chain as a space curve, the authors identify curvature (κ) and torsion (τ) as the fundamental geometric variables. These variables are treated as components of a U(1) gauge field, ensuring invariance under local rotations of the Frenet frame. By discretizing the curve onto a regular lattice, each site carries a real curvature variable and a complex phase variable that plays the role of a gauge connection. The Hamiltonian consists of three parts: (i) an Ising‑like nearest‑neighbor term representing elastic stiffness, (ii) a Chern–Simons‑type term that penalizes phase differences and mimics electromagnetic gauge dynamics, and (iii) a topological (Chern–Simons) term that introduces chirality by favoring a non‑zero total twist (the “handedness” of the chain).
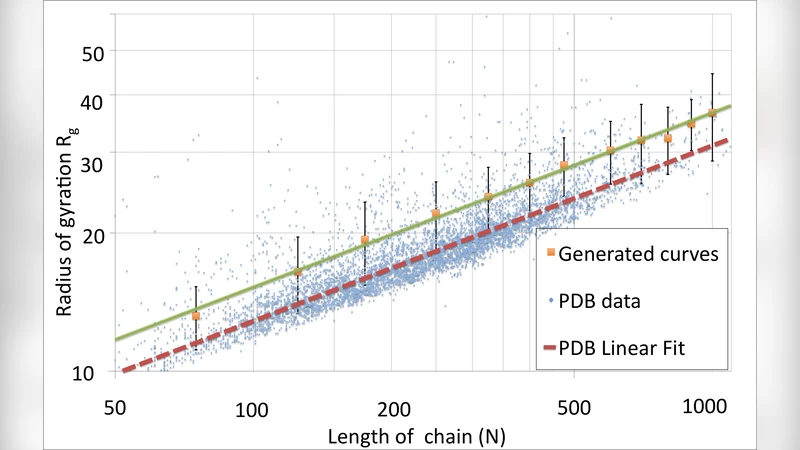
Monte‑Carlo simulations using the Metropolis algorithm explore the equilibrium configurations across a range of temperatures. As temperature decreases, the polymer collapses into compact conformations characterized by a radius‑of‑gyration scaling R_g ∝ N^ν with ν ≈ 0.33, the same compactness index observed for real proteins in the Protein Data Bank (PDB). The authors then perform a comprehensive statistical comparison between the ensembles generated by their model and a dataset of over ten thousand protein structures from the PDB. They examine (a) the R_g–N scaling, (b) contact maps (distribution of inter‑residue distances), (c) the prevalence and spatial distribution of secondary‑structure motifs such as α‑helices and β‑sheets, and (d) the distribution of the total topological twist. In all cases the model reproduces the empirical distributions with remarkable fidelity. Notably, the topological term alone is sufficient to generate realistic secondary‑structure patterns, indicating that chirality and twist are key drivers of protein‑like folding even in a homopolymer without sequence heterogeneity.
The discussion acknowledges the model’s simplifications: it neglects side‑chain specificity, branching, and explicit solvent effects. Nevertheless, the ability of a minimal gauge‑invariant framework to capture the universal statistical properties of folded proteins suggests that many aspects of protein folding belong to a broader universality class governed by geometry and topology rather than detailed chemistry. The authors propose extensions to incorporate heterogeneity, multi‑chain interactions, and applications to other biopolymers such as DNA or synthetic nanowires.
In conclusion, the study demonstrates that a gauge‑field‑theoretic lattice model, built on curvature, torsion, and a chiral topological term, can faithfully reproduce the compactness, contact statistics, and secondary‑structure content of real proteins. This work provides a new theoretical lens for understanding protein folding as a manifestation of universal geometric and topological constraints, opening avenues for efficient coarse‑grained simulations and the design of foldable synthetic polymers.
Comments & Academic Discussion
Loading comments...
Leave a Comment