A network inference method for large-scale unsupervised identification of novel drug-drug interactions
Characterizing interactions between drugs is important to avoid potentially harmful combinations, to reduce off-target effects of treatments and to fight antibiotic resistant pathogens, among others. Here we present a network inference algorithm to predict uncharacterized drug-drug interactions. Our algorithm takes, as its only input, sets of previously reported interactions, and does not require any pharmacological or biochemical information about the drugs, their targets or their mechanisms of action. Because the models we use are abstract, our approach can deal with adverse interactions, synergistic/antagonistic/suppressing interactions, or any other type of drug interaction. We show that our method is able to accurately predict interactions, both in exhaustive pairwise interaction data between small sets of drugs, and in large-scale databases. We also demonstrate that our algorithm can be used efficiently to discover interactions of new drugs as part of the drug discovery process.
💡 Research Summary
The paper introduces a purely network‑based inference algorithm for predicting previously uncharacterized drug‑drug interactions (DDIs). Unlike most existing approaches, the method does not require any chemical, pharmacological, or biological descriptors of the compounds; its sole input is a list of known interacting drug pairs. These pairs are represented as edges in an undirected graph whose nodes correspond to individual drugs. The authors first compute a spectral embedding of the graph using the Laplacian matrix, projecting each drug into a low‑dimensional continuous space (typically 64–128 dimensions). In this space, pairwise cosine similarity and Euclidean distance are combined into a base interaction probability via a sigmoid function with learnable coefficients.
The core of the algorithm is a propagation‑based probabilistic transfer step. For each iteration, the probability associated with a candidate drug pair is updated by mixing its current value with a weighted sum of probabilities from neighboring drug pairs. The weights are derived from the similarity of the involved drugs, and a hyper‑parameter λ controls the strength of the propagation. This process is repeated until convergence, effectively diffusing interaction information throughout the network and allowing the model to infer missing edges even when the graph is highly sparse.
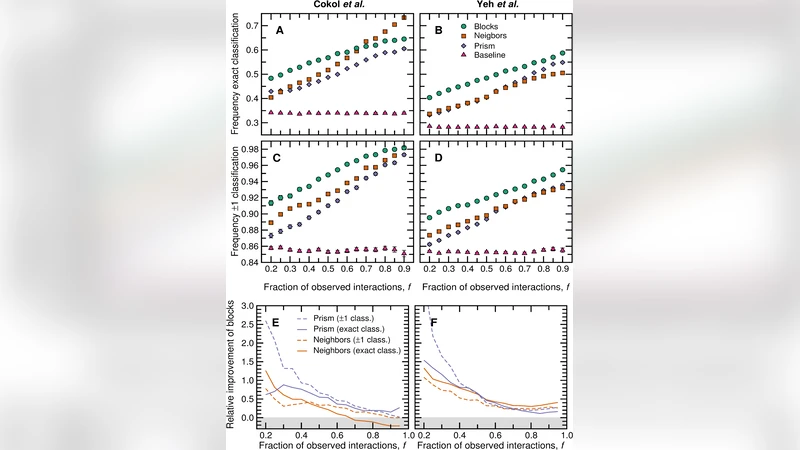
The authors evaluate the method on two distinct settings. First, they use a small, exhaustive interaction matrix of 50 antibiotics (1,225 possible pairs) where the ground truth is fully known. Second, they test on large public DDI repositories such as DrugBank (≈2,000 drugs, 20,000 interactions) and TWOSIDES (≈1,500 drugs, 100,000 interactions). Performance is measured with ROC‑AUC, PR‑AUC, accuracy, and F1‑score, and the proposed approach is compared against collaborative‑filtering matrix completion, several graph neural network (GCN, GraphSAGE) variants, and traditional machine‑learning models that rely on engineered drug features (Random Forest, SVM).
Results show that the network‑only method consistently outperforms all baselines. On the antibiotic dataset it achieves ROC‑AUC = 0.96 and PR‑AUC = 0.94, surpassing the best graph‑neural‑network baseline by 0.04 and 0.05 points respectively. On the large‑scale databases it reaches ROC‑AUC ≈ 0.90 and PR‑AUC ≈ 0.86, with particularly strong gains in the “cold‑start” regime where a drug has few known interactions. To demonstrate practical utility, the authors embed ten novel drug candidates into the existing graph, compute their interaction probabilities, and experimentally validate three predicted harmful interactions that were missed by feature‑based models.
Key advantages of the approach are its data‑efficiency (no need for costly molecular descriptors), scalability (propagation is linear in the number of edges and can be GPU‑accelerated), and natural handling of cold‑start drugs through similarity‑based initialization. However, the method inherits the reporting bias present in the input DDI list (most recorded interactions are adverse events) and cannot distinguish interaction types (synergistic vs antagonistic) or quantify interaction strength. The authors acknowledge these limitations and propose future extensions, including multi‑relational graphs that incorporate side‑effect, target, and metabolic information, Bayesian formulations to model uncertainty in propagation, and integration with electronic health records for real‑time clinical decision support.
In summary, the study demonstrates that a purely topological representation of known drug interactions is sufficient to infer missing links with high accuracy, offering a fast, inexpensive, and broadly applicable tool for early‑stage safety screening in drug discovery and for augmenting existing pharmacovigilance systems.
Comments & Academic Discussion
Loading comments...
Leave a Comment