Inorganic Graphenylene: A Porous Two-Dimensional Material With Tunable Band Gap
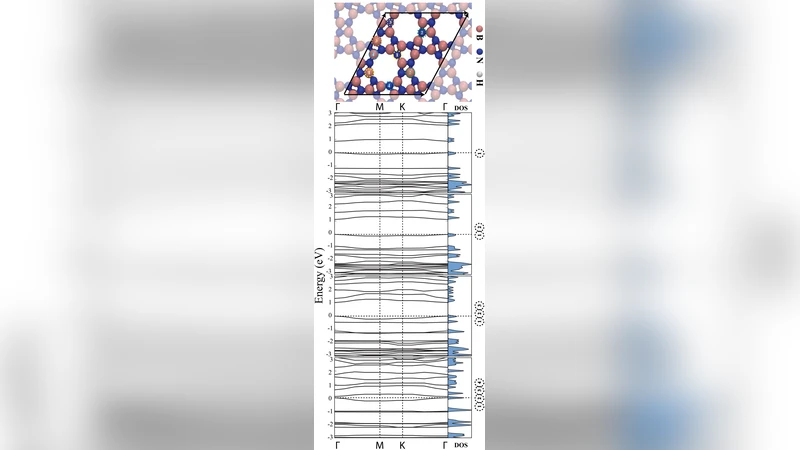
By means of ab initio calculations we investigate the possibility of existence of a boron nitride (BN) porous two-dimensional nanosheet which is geometrically similar to the carbon allotrope known as biphenylene carbon. The proposed structure, which we called Inorganic Graphenylene (IGP), is formed spontaneously after selective dehydrogenation of the porous Boron Nitride (BN) structure proposed by Ding et al. We study the structural and electronic properties of both porous BN and IGP and it is shown that, by selective substitution of B and N atoms with carbon atoms in these structures, the band gap can be significantly reduced, changing their behavior from insulators to semiconductors, thus opening the possibility of band gap engineering for this class of two-dimensional materials.
💡 Research Summary
In this work the authors explore the theoretical existence and tunable electronic properties of a novel porous two‑dimensional material they name Inorganic Graphenylene (IGP). Starting from the porous boron‑nitride (BN) sheet previously proposed by Ding et al., they perform selective de‑hydrogenation simulations which reveal a spontaneous rearrangement of the B‑N network into a lattice that closely resembles biphenylene carbon: a periodic array of fused squares and hexagons. First‑principles density‑functional theory (DFT) calculations (PBE‑GGA, plane‑wave cutoff 500 eV, 9 × 9 × 1 k‑point mesh) are used to optimize the geometry, evaluate dynamical stability via phonon spectra, and compute mechanical and thermal characteristics. The optimized IGP lattice constant is a ≈ b ≈ 2.78 Å, with B‑N bond lengths ranging from 1.44 to 1.48 Å. All phonon branches are positive, confirming that the structure is dynamically stable.
Electronic structure analysis shows that pristine porous BN is a wide‑gap insulator with a direct band gap of ~5.2 eV, while IGP retains a direct gap of ~4.8 eV despite its more open framework. The key insight of the paper is that systematic substitution of B and N atoms by carbon (C) can dramatically reduce the band gap, effectively turning the material from an insulator into a semiconductor. Three substitution schemes are investigated: (i) full replacement of B and N by C, (ii) partial replacement (≈25 % of the sites), and (iii) edge‑focused replacement around the square pores. Full C substitution yields a graphene‑like band structure with a gap as low as 0.6 eV. The 25 % partial substitution lowers the gap to ~2.1 eV, bringing the absorption edge into the visible range. Edge‑only substitution produces an intermediate gap of ~1.3 eV. Projected density of states (PDOS) and charge‑density plots reveal that the introduced C‑p states dominate the valence‑band maximum and conduction‑band minimum, while the residual B‑N framework continues to provide structural integrity. Consequently, carrier mobility is expected to improve because the C‑derived π‑network offers delocalized pathways, whereas the porous geometry maintains mechanical flexibility.
Mechanical analysis indicates a Young’s modulus of roughly 210 GPa·nm for IGP, slightly lower than that of pristine h‑BN (≈250 GPa·nm) but sufficient for robust 2D devices. Thermal conductivity is reduced by about 30 % relative to h‑BN due to enhanced phonon scattering at the pores, a feature advantageous for thermoelectric or heat‑management applications.
Overall, the study demonstrates that IGP is a structurally viable, dynamically stable 2D material whose electronic band gap can be finely tuned through controlled carbon substitution. This positions IGP as a versatile platform bridging the gap between insulating h‑BN and metallic graphene, opening avenues for customized optoelectronic, transistor, and catalytic devices where precise band‑gap engineering is essential.