A State Representation Approach for Atomistic Time-Dependent Transport Calculations in Molecular Junctions
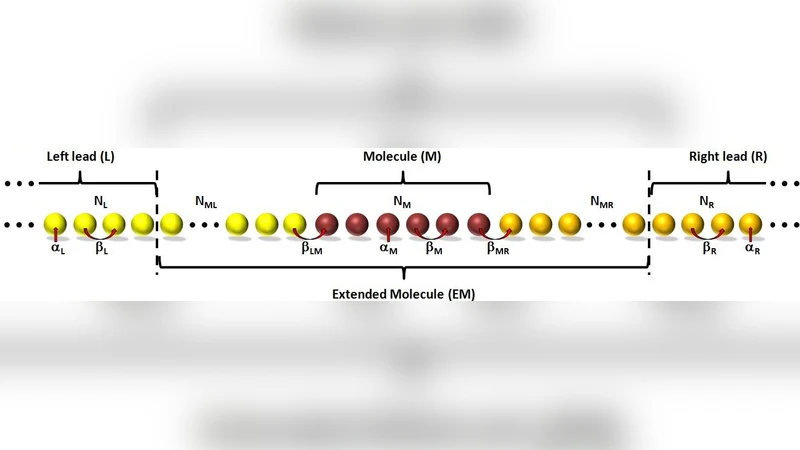
We propose a new method for simulating electron dynamics in open quantum systems out of equilibrium, using a finite atomistic model. The proposed method is motivated by the intuitive and practical nature of the driven Liouville von-Neumann equation approach of S'anchez et al. [J. Chem. Phys., 124, 214708 (2006)]. A key ingredient of our approach is a transformation of the Hamiltonian matrix from an atomistic to a state representation of the molecular junction. This allows us to uniquely define the bias voltage across the system while maintaining a proper thermal electronic distribution within the finite lead models. Furthermore, it allows us to investigate complex molecular junctions, including non-linear setups and multi-lead configurations. A heuristic derivation of our working equation leads to explicit expressions for the damping and driving terms, which serve as appropriate electron sources and sinks that effectively “open” the finite model system. Although the method does not forbid it, in practice we find neither violation of Pauli’s exclusion principles nor deviation from density matrix positivity throughout our numerical simulations of various tight-binding model systems. We believe that the new approach offers a practical and physically sound route for performing atomistic time-dependent transport calculations in realistic molecular junctions.
💡 Research Summary
The paper introduces a novel computational framework for simulating time‑dependent electron transport in open quantum systems using a finite, atomistic description of the leads and the molecular junction. Building on the driven Liouville‑von‑Neumann (DLvN) approach originally proposed by Sánchez et al., the authors address two longstanding practical issues: (i) how to impose a well‑defined bias voltage across a finite lead model, and (ii) how to maintain a proper thermal electronic distribution inside those leads without resorting to infinite reservoirs or complex self‑energy calculations.
The key methodological advance is a transformation of the Hamiltonian from the conventional atomic orbital basis to a “state representation” in which each lead and the central molecule are expressed in terms of their own eigenstates. In this representation the bias voltage can be assigned unambiguously to the lead states, while the occupation of each state is forced toward a Fermi‑Dirac distribution at the prescribed temperature and chemical potential. To emulate the effect of an open system, the authors augment the Liouville‑von‑Neumann equation with two additional terms: a driving term that continuously pumps the lead states toward their target Fermi‑Dirac occupations, and a damping term that absorbs electrons leaving the central region into the leads (or injects them from the leads into the central region). These terms are derived heuristically, yielding explicit expressions for the corresponding rates (γ‑drive and γ‑damp) that are directly linked to the lead density of states and the chosen bias.
The resulting equation of motion reads
(\dot{\rho}= -\frac{i}{\hbar}