Statistical-mechanical lattice models for protein-DNA binding in chromatin
Statistical-mechanical lattice models for protein-DNA binding are well established as a method to describe complex ligand binding equilibriums measured in vitro with purified DNA and protein components. Recently, a new field of applications has opened up for this approach since it has become possible to experimentally quantify genome-wide protein occupancies in relation to the DNA sequence. In particular, the organization of the eukaryotic genome by histone proteins into a nucleoprotein complex termed chromatin has been recognized as a key parameter that controls the access of transcription factors to the DNA sequence. New approaches have to be developed to derive statistical mechanical lattice descriptions of chromatin-associated protein-DNA interactions. Here, we present the theoretical framework for lattice models of histone-DNA interactions in chromatin and investigate the (competitive) DNA binding of other chromosomal proteins and transcription factors. The results have a number of applications for quantitative models for the regulation of gene expression.
💡 Research Summary
The paper presents a comprehensive statistical‑mechanical lattice framework for describing protein‑DNA interactions within chromatin, extending classic lattice models that have traditionally been applied to purified in‑vitro systems. The authors begin by treating the DNA molecule as a one‑dimensional lattice of binding sites. Each lattice unit can exist in one of several states: empty, occupied by a histone octamer (or a specific histone modification), or occupied by a transcription factor (TF) or other chromatin‑associated protein. The model incorporates the intrinsic binding free energies (ΔG) and dissociation constants (Kd) for each protein type, as well as steric exclusion that prevents overlapping occupancy.
In the first part, a “single‑component” lattice model is constructed for histone‑DNA binding. Histones are modeled as covering ~147 bp, which translates into a block of consecutive lattice sites that become unavailable for other ligands once a histone is placed. The partition function of the entire genome is derived using a transfer‑matrix formalism, where the matrix elements encode the statistical weights of transitions between empty and histone‑occupied states, as well as possible cooperative or anti‑cooperative interactions between neighboring nucleosomes. By diagonalizing the transfer matrix, the dominant eigenvalue yields the free‑energy density, and the associated eigenvector provides the probability that any given site is occupied by a nucleosome.
The second, more elaborate component introduces competing TFs and other chromatin proteins. TFs can bind only to sites not already covered by nucleosomes, and each TF possesses a sequence‑specific binding constant that reflects its DNA motif affinity. The model also allows for “cooperative” terms that capture how a particular histone modification (e.g., H3K27ac) can modulate TF affinity, as well as “anti‑cooperative” terms that represent pure steric blockage. The transfer matrix is expanded to three‑state (empty, nucleosome, TF) form, and the same eigenvalue analysis yields genome‑wide occupancy probabilities for both nucleosomes and TFs.
To validate the analytical approach, the authors perform Monte‑Carlo simulations of the lattice dynamics, confirming that the equilibrium distributions obtained from the transfer‑matrix solution match the stochastic results. They then apply the framework to real genome‑wide data: ChIP‑seq profiles of a histone modification (H3K27ac) and a TF (NF‑κB) in human cells. By fitting the model parameters (ΔG for nucleosome binding, ΔG_coop for modification‑dependent TF affinity, and Kd for TF‑DNA binding) to the observed ChIP‑seq signal, the model reproduces key features of the data. In regions of high nucleosome density, TF occupancy is strongly suppressed, whereas in domains where the activating histone mark is enriched, the model predicts a sharp increase in TF binding—a “bypass” effect that cannot be captured by a simple exclusion model.
The paper’s contributions are threefold. First, it provides a unified statistical‑mechanical description that simultaneously accounts for nucleosome positioning, histone modifications, and TF competition. Second, the transfer‑matrix method delivers a computationally efficient solution that scales to whole‑genome analyses, making it feasible to integrate large‑scale epigenomic datasets. Third, the framework enables inverse inference of biophysical parameters from genome‑wide occupancy data, thereby linking measurable ChIP‑seq signals to underlying free‑energy landscapes and cooperative interactions.
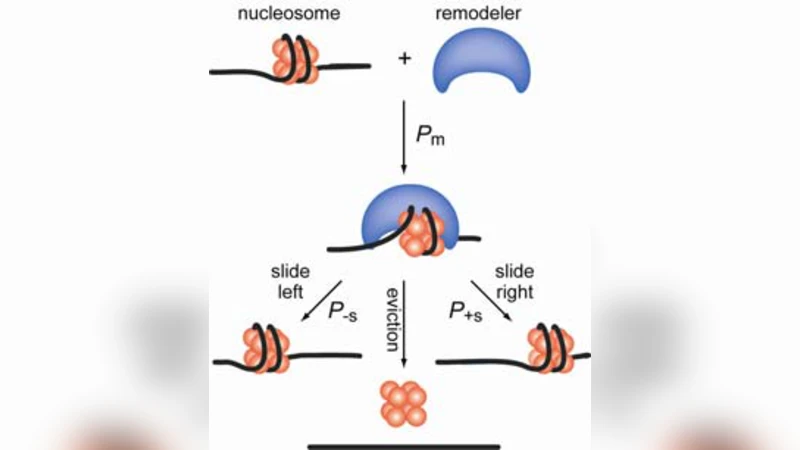
The authors discuss several future directions. Extending the lattice to incorporate DNA methylation, nucleosome remodelers, and higher‑order chromatin architecture (loops, topologically associating domains) would increase biological realism. Incorporating kinetic rates for binding and unbinding would allow the model to address dynamic processes such as TF search kinetics and nucleosome turnover. Finally, the approach could be employed to predict the impact of pharmacological agents that target histone‑modifying enzymes or chromatin remodelers, offering a quantitative tool for drug discovery in epigenetic therapy.
Comments & Academic Discussion
Loading comments...
Leave a Comment