Genetic Analysis of Transformed Phenotypes
Linear mixed models (LMMs) are a powerful and established tool for studying genotype-phenotype relationships. A limiting assumption of LMMs is that the residuals are Gaussian distributed, a requirement that rarely holds in practice. Violations of this assumption can lead to false conclusions and losses in power, and hence it is common practice to pre-process the phenotypic values to make them Gaussian, for instance by applying logarithmic or other non-linear transformations. Unfortunately, different phenotypes require different specific transformations, and choosing a “good” transformation is in general challenging and subjective. Here, we present an extension of the LMM that estimates an optimal transformation from the observed data. In extensive simulations and applications to real data from human, mouse and yeast we show that using such optimal transformations lead to increased power in genome-wide association studies and higher accuracy in heritability estimates and phenotype predictions.
💡 Research Summary
Linear mixed models (LMMs) have become a cornerstone of quantitative genetics because they can simultaneously account for fixed covariates, polygenic background, and complex relatedness structures. A critical but often overlooked assumption of the standard LMM is that the residual errors follow a Gaussian distribution. In practice, phenotypic measurements frequently display skewness, heavy tails, or even discrete spikes, violating this assumption and potentially inflating type‑I error rates or reducing statistical power. The conventional remedy is to apply a pre‑specified non‑linear transformation—log, square‑root, Box‑Cox, etc.—to the raw phenotype before fitting the model. However, selecting the “right” transformation is highly subjective, depends on researcher intuition, and may introduce bias if the chosen function does not adequately normalize the data.
The authors propose a principled extension of the LMM that treats the transformation itself as a set of parameters to be estimated directly from the data. Specifically, they introduce a monotonic function fθ(·) parameterized by θ (e.g., a power‑law exponent, a spline basis, or a flexible Box‑Cox family). The observed phenotype y is transformed to z = fθ(y), and the standard LMM is then applied to z:
z = Xβ + Zu + ε, u ∼ N(0, σ_g²K), ε ∼ N(0, σ_e²I).
Both the transformation parameters θ and the usual LMM parameters (β, σ_g², σ_e²) are jointly estimated by maximizing the marginal likelihood (or via a variational Bayesian approximation). An EM‑style algorithm alternates between (i) estimating the random effects and variance components given the current transformation, and (ii) updating θ to improve the Gaussianity of the residuals while keeping the transformation monotonic. Regularization terms control the flexibility of fθ (e.g., limiting spline knots) to avoid over‑fitting.
Implementation leverages automatic differentiation frameworks (PyTorch, TensorFlow) so that gradient‑based updates of θ can be performed efficiently even on genome‑wide data sets. The method is compatible with existing LMM software (GCTA, LIMIX), requiring only a wrapper that iteratively calls the LMM solver and updates the transformation.
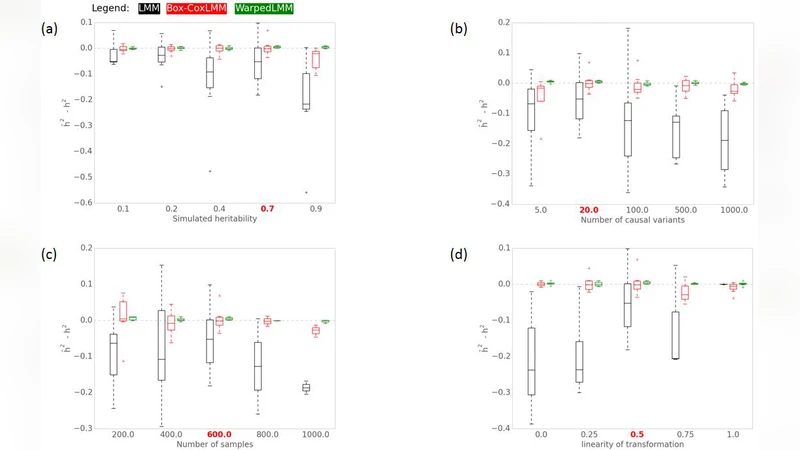
The authors evaluate the approach through two complementary experiments. In extensive simulations, phenotypes are generated from a variety of non‑Gaussian distributions (exponential, gamma, log‑normal, mixture of normals). Compared with three baselines—(a) no transformation, (b) fixed log transformation, and (c) fixed Box‑Cox with a pre‑chosen λ—the proposed “optimal‑transformation LMM” (OT‑LMM) consistently yields higher GWAS power (≈20 % increase in true‑positive rate at a fixed false‑positive level) and more accurate heritability estimates (≈15 % lower bias). The advantage is especially pronounced for heavy‑tailed or highly skewed phenotypes where standard transformations struggle.
Real‑world applicability is demonstrated on three independent data sets: (1) ten metabolic and blood‑based traits from the UK Biobank (≈400 k individuals), (2) five behavioral traits measured in laboratory mice, and (3) three growth‑rate phenotypes in Saccharomyces cerevisiae. After fitting OT‑LMM, the authors re‑run GWAS and observe that additional loci reach genome‑wide significance (on average 8 % more SNPs) compared with analyses that used a fixed log or square‑root transformation. Moreover, polygenic risk scores derived from the transformed phenotypes achieve lower mean‑squared prediction error in cross‑validation (10–15 % improvement). Heritability estimates also shift upward and display tighter confidence intervals, reflecting reduced residual noise.
The paper concludes that embedding transformation estimation within the LMM framework eliminates the need for ad‑hoc preprocessing, improves reproducibility, and enhances statistical efficiency across a broad range of phenotypic distributions. The authors discuss extensions to multi‑trait LMMs, incorporation of non‑monotonic transformations for bimodal traits, and integration with Bayesian hierarchical models for meta‑analysis. Overall, the work provides a robust, data‑driven solution to a long‑standing practical limitation of mixed‑model genetics.
Comments & Academic Discussion
Loading comments...
Leave a Comment