A stochastic model of catalytic reaction networks in protocells
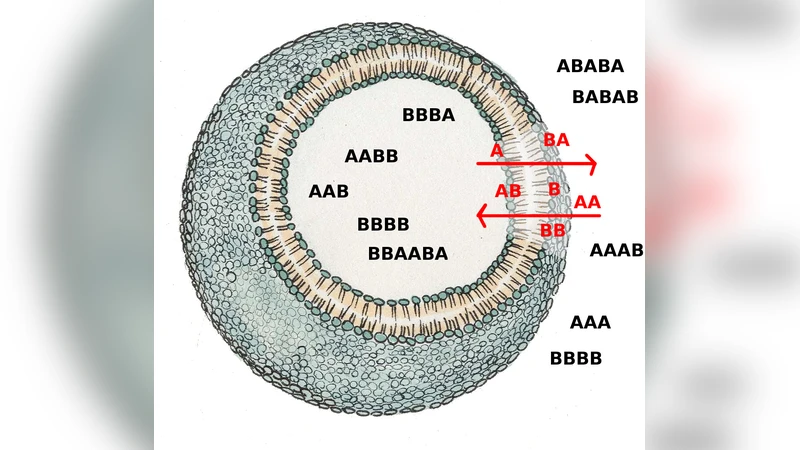
Protocells are supposed to have played a key role in the self-organizing processes leading to the emergence of life. Existing models either (i) describe protocell architecture and dynamics, given the existence of sets of collectively self-replicating molecules for granted, or (ii) describe the emergence of the aforementioned sets from an ensemble of random molecules in a simple experimental setting (e.g. a closed system or a steady-state flow reactor) that does not properly describe a protocell. In this paper we present a model that goes beyond these limitations by describing the dynamics of sets of replicating molecules within a lipid vesicle. We adopt the simplest possible protocell architecture, by considering a semi-permeable membrane that selects the molecular types that are allowed to enter or exit the protocell and by assuming that the reactions take place in the aqueous phase in the internal compartment. As a first approximation, we ignore the protocell growth and division dynamics. The behavior of catalytic reaction networks is then simulated by means of a stochastic model that accounts for the creation and the extinction of species and reactions. While this is not yet an exhaustive protocell model, it already provides clues regarding some processes that are relevant for understanding the conditions that can enable a population of protocells to undergo evolution and selection.
💡 Research Summary
This paper addresses a central gap in origin‑of‑life modeling by coupling stochastic catalytic reaction networks (CRSs) with a minimal protocell architecture. Existing approaches either assume pre‑existing self‑replicating molecular sets within a compartment, or study the emergence of such sets in open‑flow reactors (CSTRs) that lack the defining features of a protocell—namely a semi‑permeable membrane, finite volume, and the possibility of growth and division. The authors therefore construct a simplified protocell model: a spherical lipid vesicle with a fixed volume, a membrane that allows only molecules shorter than a prescribed length to pass freely, and instantaneous equilibration of permeable species across the membrane. Growth and division are deliberately omitted to focus on short‑time dynamics.
Molecular species are represented as strings of letters (bricks) drawn from a finite alphabet, with monomers (single bricks) and polymers (longer strings). Two elementary reactions are defined: cleavage (splitting a polymer into two shorter fragments) and condensation (joining two polymers into a longer one). Crucially, every reaction requires a catalyst; only polymers exceeding a minimum length can act as catalysts, and a catalyst may accelerate multiple reactions while a reaction may have several catalysts. This catalytic constraint mirrors the high activation energy of uncatalyzed chemistry and implements the concept of autocatalytic sets (ACSs).
The dynamics are simulated using Gillespie’s stochastic simulation algorithm, which naturally captures discrete events such as the birth and extinction of species and the stochastic growth or shrinkage of the reaction graph—features that deterministic ODE models miss. Because the vesicle volume is tiny (on the order of 10⁻³–1 µm³), the absolute number of molecules for many species is in the tens or even single digits, making stochastic fluctuations dominant.
A series of computational experiments explores three axes of variability: (1) different random initial concentrations drawn from the same distribution, (2) different random orders of reaction events (path dependency), and (3) different underlying chemistries (e.g., varying the minimum catalyst length or reaction rates, analogous to RAF sets). The authors find that the semi‑permeable membrane dramatically increases heterogeneity among protocells: even with identical external conditions, the internal composition diverges because only a subset of molecules can cross the membrane, and stochastic early appearance of a catalyst can lock a protocell into a particular reaction trajectory. When a catalyst emerges early, it can seed an ACS that rapidly dominates the internal chemistry; if it appears later, alternative pathways may prevail or the network may collapse. Changing the catalytic rules alters the frequency and stability of ACS formation, suggesting that specific chemical environments could have favored the emergence of self‑sustaining networks.
The paper also discusses limitations. The fixed‑volume, instantaneous‑equilibrium membrane assumption neglects osmotic pressure, membrane growth, and the feedback between internal chemistry and vesicle division—processes essential for Darwinian evolution. Moreover, the model permits unbounded concentration growth for non‑permeable species, which is non‑physical beyond short simulation windows. The authors propose future extensions that relax these simplifications, incorporating vesicle growth, division, and more realistic transport kinetics.
In summary, this work provides the first quantitative framework that embeds stochastic catalytic reaction networks within a protocell‑like compartment. It demonstrates how compartmentalization, membrane selectivity, and stochasticity together can generate diverse chemical trajectories and facilitate the spontaneous emergence of autocatalytic sets, offering a plausible mechanistic bridge between chemistry and early cellular evolution.
Comments & Academic Discussion
Loading comments...
Leave a Comment