Competition driven cancer immunoediting
It is a well-established fact that tumors up-regulate glucose consumption to meet increasing demands for rapidly available energy by switching to purely glycolytic mode of glucose metabolism. What is often neglected is that cytotoxic cells of the immune system also have increased energy demands and also switch to pure glycolysis when they are in an activated state. Moreover, while cancer cells can revert back to aerobic metabolism, rapidly proliferating cytotoxic lymphocytes are incapable of performing their function when adequate resources are lacking. Consequently, in the tumor microenvironment there must exist competition for the common resources between cancer cells and the cells of the immune system, which may drive a lot of the tumor-immune dynamics. Proposed here is a model of tumor-immune-glucose interactions, formulated as a predator-prey-common resource type system, which allows to investigate possible dynamical behaviors that may arise as a result of competition for glucose, including tumor elimination, tumor dormancy and unrestrained tumor growth.
💡 Research Summary
The manuscript “Competition driven cancer immunoediting” proposes a novel mechanistic framework that places metabolic competition for glucose at the heart of tumor‑immune dynamics. Building on the well‑established observations that (i) rapidly proliferating cancer cells adopt a glycolytic phenotype (the Warburg effect) to satisfy their high ATP and biosynthetic demands, and (ii) activated cytotoxic lymphocytes (CTLs, NK cells) likewise switch to aerobic glycolysis to fuel effector functions, the authors argue that both cell populations draw from the same limited pool of extracellular glucose within the tumor microenvironment (TME).
To formalize this intuition, the authors construct a predator‑prey‑common‑resource (PPCR) model consisting of three coupled ordinary differential equations: (1) tumor cell density N(t), (2) immune effector density I(t), and (3) glucose concentration G(t). Tumor growth is modeled as a glucose‑dependent logistic term with a consumption coefficient α; immune activation is driven by tumor‑cell encounter but is attenuated by a glucose‑dependent death term δ(G) that sharply rises when G falls below a critical threshold. Glucose dynamics incorporate a constant influx S (representing vascular supply) and depletion terms αN and βI, where β denotes the per‑cell glucose uptake of activated immune cells. The model therefore captures three essential processes: (a) resource inflow, (b) resource consumption by two competing consumers, and (c) predator‑prey interaction mediated by immune killing.
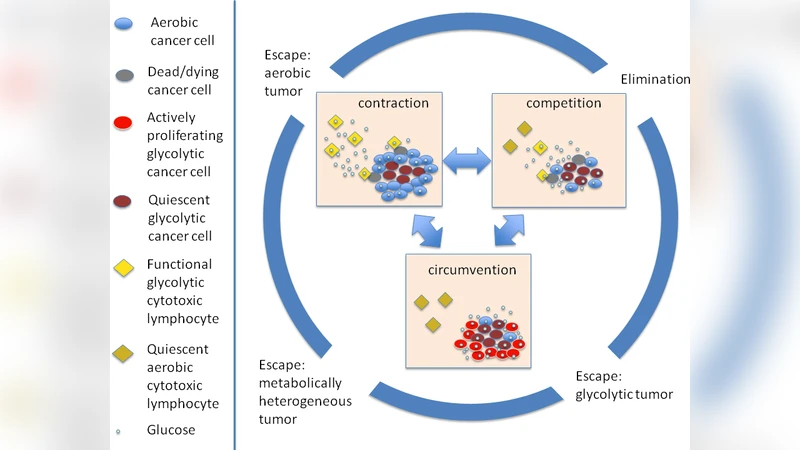
Analytical fixed‑point analysis reveals three biologically meaningful equilibria: (i) a tumor‑free state (N≈0, I>0, G>0) corresponding to successful immune eradication; (ii) a dormant or “equilibrium” state (N>0, I>0, G≈G*), where glucose limitation forces a stalemate and tumor size stabilizes; and (iii) an uncontrolled growth state (N≫I, G≈0) in which immune cells are starved of glucose, lose cytotoxic function, and the tumor expands unchecked. Bifurcation analysis shows that the transition between these regimes is governed primarily by the glucose supply rate S, the relative consumption rates α/β, and the steepness of the immune death function δ(G). Small variations in any of these parameters can shift the system across a transcritical bifurcation, turning a dormant tumor into either eradication or aggressive progression.
Numerical simulations, initialized under tumor‑dominant, immune‑dominant, and balanced conditions, reproduce the three outcomes and illustrate dynamic trajectories such as: (a) rapid tumor clearance when glucose is abundant and immune killing is efficient; (b) prolonged dormancy when glucose is modestly limited, producing a “steady‑state” tumor burden; and (c) runaway tumor growth when glucose is either oversupplied (fueling cancer cells) or severely restricted (crippling immune cells). The authors highlight that the “equilibrium” phase of classic cancer immunoediting (elimination → equilibrium → escape) can be interpreted as a metabolic balance point, where competition for glucose determines whether the immune system can maintain control.
Importantly, the paper links these theoretical insights to therapeutic implications. First, it suggests that metabolic adjuvants—such as GLUT1 inhibitors, glycolysis blockers, or dietary glucose restriction—could tip the competition in favor of immune cells, enhancing checkpoint blockade or adoptive cell therapy. Second, it proposes re‑programming immune cells to rely less on glucose (e.g., by promoting oxidative phosphorylation or fatty‑acid oxidation) to render them more resilient in a glucose‑poor TME. The authors cite experimental evidence that high systemic glucose or tumor‑derived lactate impairs T‑cell function, supporting the model’s predictions.
In the discussion, the authors acknowledge limitations: the model treats glucose as a single homogeneous resource, ignores spatial heterogeneity, and does not incorporate other metabolites (glutamine, lactate) or immunosuppressive factors (PD‑L1, Tregs). They propose extensions such as spatial reaction‑diffusion models, inclusion of additional nutrients, and coupling to angiogenesis dynamics.
Overall, the manuscript delivers a concise yet powerful quantitative framework that integrates tumor metabolism, immune effector biology, and resource competition. By demonstrating how glucose scarcity can drive immunoediting outcomes ranging from elimination to escape, it opens new avenues for combined metabolic‑immune therapies and underscores the need for experimental validation of glucose‑focused interventions in cancer treatment.
Comments & Academic Discussion
Loading comments...
Leave a Comment