Inverse Quantum Chemistry: Concepts and Strategies for Rational Compound Design
The rational design of molecules and materials is becoming more and more important. With the advent of powerful computer systems and sophisticated algorithms, quantum chemistry plays an important role in rational design. While traditional quantum chemical approaches predict the properties of a predefined molecular structure, the goal of inverse quantum chemistry is to find a structure featuring one or more desired properties. Herein, we review inverse quantum chemical approaches proposed so far and discuss their advantages as well as their weaknesses.
💡 Research Summary
The paper “Inverse Quantum Chemistry: Concepts and Strategies for Rational Compound Design” provides a comprehensive review of the emerging field of inverse quantum chemistry, which seeks to determine molecular structures that exhibit predefined properties rather than predicting properties from known structures. The authors begin by outlining the limitations of conventional forward quantum‑chemical approaches, which require exhaustive screening of vast chemical spaces—estimated to contain between 10²⁰ and 10⁶⁰ possible molecules—to locate a candidate with a desired property. They argue that a true inverse methodology, which directly maps target properties onto molecular architectures, would dramatically reduce computational cost and accelerate rational design in chemistry, materials science, and related disciplines.
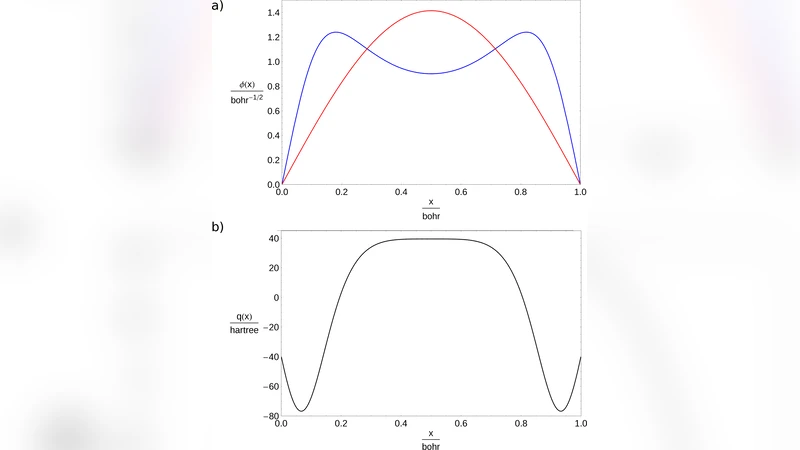
The review is organized into several thematic sections. Section 2 introduces inverse spectral theory, tracing its origins to the work of Ambarzumian and Borg. The authors explain that reconstructing a Hamiltonian from a single set of eigenvalues is generally impossible; a complementary spectrum (e.g., different boundary conditions) is required to uniquely determine the underlying potential. This mathematical insight underpins the non‑uniqueness problem in chemistry: many distinct molecules can share similar observable properties.
Section 3 focuses on Inverse Perturbation Analysis (IPA), a method pioneered by Kosman and Hinze. Starting from an approximate potential (often a Rydberg–Klein–Rees form), the exact potential is expressed as the sum of the approximation and a small correction. By comparing experimentally measured energy levels with those computed from the approximate potential, the correction is obtained via first‑order perturbation theory and expanded in a chosen basis set. The authors discuss practical implementations, highlighting the sensitivity of results to the choice of basis functions (global polynomials versus local Gaussians) and the ill‑conditioned nature of the inverse problem. Recent advances using singular‑value decomposition to stabilize the solution are noted.
Section 4 examines model‑equation approaches and wave‑function optimization. Here the wavefunction itself becomes a variational variable, and target properties such as dipole moments, reaction barriers, or excitation energies are directly minimized. The authors describe how machine‑learning‑based parameterizations can be combined with traditional variational principles to navigate the high‑dimensional space of possible wavefunctions.
Section 5 surveys modern sampling and optimization techniques—genetic algorithms, Bayesian optimization, reinforcement learning—that have been applied to the inverse design of molecules. These methods efficiently explore the combinatorial chemical space while steering toward structures that satisfy the predefined property constraints.
Section 6 discusses the integration of quantitative structure‑activity relationship (QSAR) models into inverse design. Large experimental databases are used to train regression or classification models; the trained models are then inverted (often via generative neural networks) to propose candidate structures that are predicted to meet the target property. Subsequent quantum‑chemical validation closes the loop.
Sections 7.1 and 7.2 extend the discussion to solid‑state materials, describing inverse strategies for tailoring band structures, conductivity, and optical responses. Section 7.3 introduces linear combinations of atom‑centered potentials (ACP) as a continuous representation of atomic types and positions, enabling gradient‑based optimization of crystalline structures. Section 7.4 presents alchemical potentials, which treat atomic transmutations as continuous parameter changes, allowing the calculation of property derivatives with respect to elemental composition.
Sections 8 and 9 showcase the authors’ own contributions, where they combine several of the reviewed techniques—IPA, ACP, and alchemical derivatives—to design catalysts and photovoltaic materials with improved performance.
In the concluding section, the authors emphasize that inverse quantum chemistry holds the promise of turning property specifications into concrete molecular designs, thereby bypassing the costly forward screening paradigm. However, they also stress fundamental challenges: the non‑uniqueness of solutions, the ill‑posed nature of high‑dimensional inverse problems, and the need for robust, scalable optimization algorithms that can incorporate experimental feedback. Future research directions include developing stronger mathematical foundations for inverse problems, improving numerical stability through advanced regularization techniques, and creating integrated workflows that couple high‑throughput quantum calculations with data‑driven models. By addressing these issues, inverse quantum chemistry could become a cornerstone of rational design across chemistry, materials science, and related fields.
Comments & Academic Discussion
Loading comments...
Leave a Comment