Evolution and Controllability of Cancer Networks: a Boolean Perspective
Cancer forms a robust system and progresses as stages over time typically with increasing aggressiveness and worsening prognosis. Characterizing these stages and identifying the genes driving transitions between them is critical to understand cancer progression and to develop effective anti-cancer therapies. Here, we propose a novel model of the ‘cancer system’ as a Boolean state space in which a Boolean network, built from protein interaction and gene-expression data from different stages of cancer, transits between Boolean satisfiability states by “editing” interactions and “flipping” genes. The application of our model (called BoolSpace) on three case studies - pancreatic and breast tumours in human and post spinal-cord injury in rats - reveals valuable insights into the phenomenon of cancer progression. In particular, we notice that several of the genes flipped are serine/threonine kinases which act as natural cellular switches and that different sets of genes are flipped during the initial and final stages indicating a pattern to tumour progression. We hypothesize that robustness of cancer partly stems from “passing of the baton” between genes at different stages, and therefore an effective therapy should target a “cover set” of these genes. A C/C++ implementation of BoolSpace is freely available at: http://www.bioinformatics.org.au/tools-data
💡 Research Summary
The paper introduces a novel systems‑biology framework, BoolSpace, that models cancer progression as a Boolean state space derived from protein‑protein interaction (PPI) networks and stage‑specific gene‑expression data. The authors first construct a high‑confidence human PPI backbone (≈29,600 interactions among 5,824 proteins) and, for each condition (normal, tumor, or later disease stage), compute Pearson correlation coefficients for each interacting gene pair across multiple samples. Interactions with strong positive correlation (≥ δ) are encoded as NOT‑XOR (¯⊗) clauses, while strong negative correlations (≤ ‑δ) become XOR (⊗) clauses, yielding a condition‑specific Boolean network BΩ where each gene is a binary variable and each edge is a logical clause reflecting co‑expression.
Transitions between successive conditions Ω and Ψ are captured by three types of edits to the interaction set: loss (correlation drops into the indeterminate zone), gain (correlation emerges), and toggling (sign flips). Toggling changes the logical operator of an edge from ¯⊗ to ⊗ or vice‑versa, representing a reversal of co‑expression. The set of edited edges EΩΨ thus defines how the Boolean network is rewired when moving from one stage to the next.
The central computational problem, MIN‑FLIP, asks for the smallest subset of genes whose Boolean values must be flipped (0↔1) so that the original satisfying assignment B(BΩ) remains a satisfying assignment after the edits EΩΨ are applied. In other words, MIN‑FLIP identifies the minimal driver genes that can rescue network feasibility after a stage transition. The authors prove that MIN‑FLIP is equivalent to the MIN‑ONES‑2SAT problem, which is polynomial‑time solvable for networks containing only ¯⊗/⊗ clauses but NP‑complete in the general case. To handle realistic, heterogeneous networks, they develop a fixed‑parameter tractable (FPT) algorithm parameterized by the number k of flipped genes. The algorithm runs in O(f(k)·n^c) time, where f(k) is exponential in k but independent of the total number of genes n, making it practical when k is small.
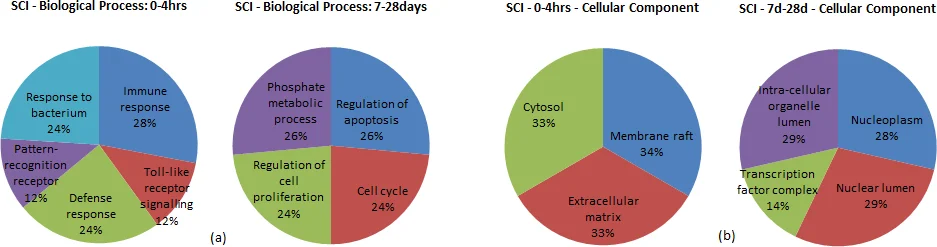
The framework is evaluated on three case studies: (1) pancreatic ductal adenocarcinoma (PDAC) using 39 paired normal‑tumor samples; (2) breast cancer subtypes; and (3) a rat spinal‑cord injury model. In the PDAC analysis, 8,701 highly correlated interactions present in normal tissue disappear in tumors, indicating loss of “accelerator” edges and “brake” edges. Two interactions (RBPMS‑RHOXF2 and SMN1‑TMSB4X) exhibit extreme correlation jumps, highlighting potential driver events. MIN‑FLIP identifies serine/threonine kinases (e.g., CDK1, MAPK1) as the primary flipped genes during early transitions, while later transitions involve DNA‑damage‑response genes (e.g., ATM, BRCA1). Similar patterns emerge in breast cancer, where different kinase and cell‑cycle regulators dominate early versus late stage flips, and in the spinal‑cord injury model, where inflammation and neuro‑regeneration pathways are rewired over time.
These results support two key biological insights: (i) cancer robustness stems partly from a “baton‑passing” mechanism where distinct functional gene sets dominate successive stages, allowing the tumor to evade therapies targeting a single pathway; and (ii) effective therapeutic strategies should aim at a “cover set” of driver genes that collectively span all stages, rather than focusing on stage‑specific single targets.
The authors release a C/C++ implementation of BoolSpace (available at the provided URL) and suggest future extensions to incorporate additional omics layers (e.g., methylation, phosphoproteomics) and larger patient cohorts. By uniting Boolean satisfiability, network editing, and fixed‑parameter algorithmics, the study offers a rigorous quantitative tool for dissecting the dynamic controllability of cancer networks and for guiding multi‑target drug design.
Comments & Academic Discussion
Loading comments...
Leave a Comment