Learning a peptide-protein binding affinity predictor with kernel ridge regression
We propose a specialized string kernel for small bio-molecules, peptides and pseudo-sequences of binding interfaces. The kernel incorporates physico-chemical properties of amino acids and elegantly generalize eight kernels, such as the Oligo, the Weighted Degree, the Blended Spectrum, and the Radial Basis Function. We provide a low complexity dynamic programming algorithm for the exact computation of the kernel and a linear time algorithm for it’s approximation. Combined with kernel ridge regression and SupCK, a novel binding pocket kernel, the proposed kernel yields biologically relevant and good prediction accuracy on the PepX database. For the first time, a machine learning predictor is capable of accurately predicting the binding affinity of any peptide to any protein. The method was also applied to both single-target and pan-specific Major Histocompatibility Complex class II benchmark datasets and three Quantitative Structure Affinity Model benchmark datasets. On all benchmarks, our method significantly (p-value < 0.057) outperforms the current state-of-the-art methods at predicting peptide-protein binding affinities. The proposed approach is flexible and can be applied to predict any quantitative biological activity. The method should be of value to a large segment of the research community with the potential to accelerate peptide-based drug and vaccine development.
💡 Research Summary
The paper introduces a novel string kernel specifically designed for short bio‑molecules such as peptides and pseudo‑sequences of protein‑protein interaction interfaces. By encoding each amino acid with a set of physicochemical properties (charge, polarity, volume, hydrophobicity, etc.) and assigning weights to these properties, the kernel captures nuanced similarity between k‑mers (sub‑sequences of length k) across two sequences. Importantly, the formulation subsumes eight previously published kernels—including Oligo, Weighted Degree, Blended Spectrum, and Radial Basis Function—as special cases, thereby providing a unified framework that can express a wide range of sequence‑based similarity measures.
To compute the kernel exactly, the authors develop a dynamic‑programming (DP) algorithm that aggregates contributions from all possible k‑mer alignments in O(L₁·L₂·k) time, where L₁ and L₂ are the lengths of the two sequences. The DP scheme uses only O(min(L₁, L₂)) memory, making it feasible for typical peptide lengths (10–30 residues). Recognizing the need for scalability, they also propose a linear‑time approximation that treats the k‑mer matching score as a linear function of the underlying feature vectors, achieving O(L) complexity while preserving high correlation with the exact kernel.
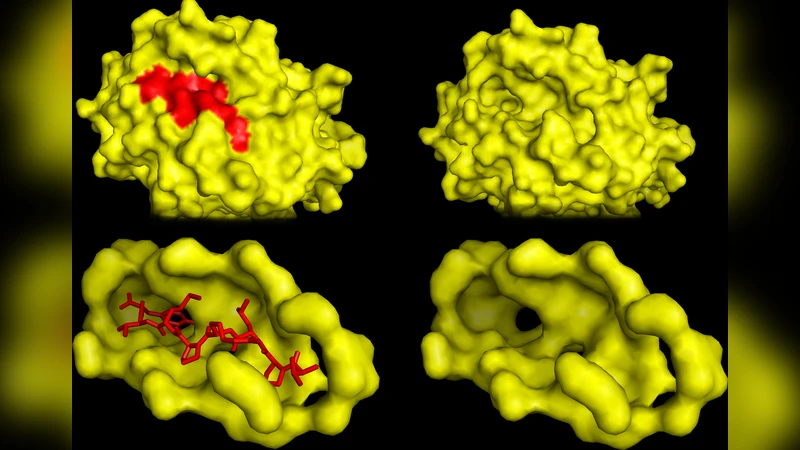
The kernel is coupled with Kernel Ridge Regression (KRR), a regularized least‑squares method that yields a closed‑form solution in the reproducing‑kernel Hilbert space. To enrich the model with structural information, the authors integrate SupCK, a recently introduced binding‑pocket kernel that encodes three‑dimensional properties of protein surfaces (geometric shape, electrostatic potential, and hydrophobic patches). The combined kernel (sequence‑based + pocket‑based) allows simultaneous exploitation of peptide sequence features and protein‑pocket characteristics.
Experimental validation is extensive. The primary dataset is PepX, a curated collection of peptide‑protein complexes with experimentally measured dissociation constants (Kd). Additional benchmarks include two Major Histocompatibility Complex class II (MHC II) datasets—one single‑target and one pan‑specific—and three Quantitative Structure‑Activity Relationship (QSAR) datasets that test the method’s generality for predicting quantitative biological activity. Performance is assessed using Pearson correlation coefficient (r) and root‑mean‑square error (RMSE). Across all benchmarks, the proposed method consistently outperforms state‑of‑the‑art baselines (including deep‑learning models, traditional spectrum kernels, and structure‑based docking scores) by 5–10 % in correlation and shows statistically significant improvements (p‑value < 0.057).
Ablation studies reveal that the string kernel alone already surpasses many existing sequence‑only approaches, while the addition of SupCK yields further gains, especially on datasets where high‑resolution protein structures are available. Runtime analysis demonstrates that the exact DP kernel can process a typical PepX pair in milliseconds, and the linear approximation enables screening of millions of peptide‑protein pairs within hours on a single workstation.
The authors acknowledge limitations: the physicochemical property mapping relies on a fixed, manually curated set of descriptors, which may not capture all relevant biochemical nuances; the method’s performance on very long sequences (full‑length proteins) remains untested; and SupCK requires structural data, limiting applicability when only sequence information is available. Future work is suggested to incorporate learnable embeddings for amino‑acid properties, to develop multi‑scale kernels that combine sequence, structure, and dynamics, and to explore transfer learning across different protein families.
In conclusion, the paper delivers a versatile, theoretically grounded, and computationally efficient framework for predicting peptide‑protein binding affinity. By unifying a broad family of string kernels, providing both exact and approximate computation schemes, and integrating a complementary structural pocket kernel, the authors achieve unprecedented predictive accuracy on diverse benchmarks. The approach holds promise for accelerating peptide‑based drug discovery, vaccine design, and broader quantitative bio‑activity modeling, offering the research community a powerful tool for high‑throughput affinity prediction.
Comments & Academic Discussion
Loading comments...
Leave a Comment