A novel canonical dual computational approach for prion AGAAAAGA amyloid fibril molecular modeling
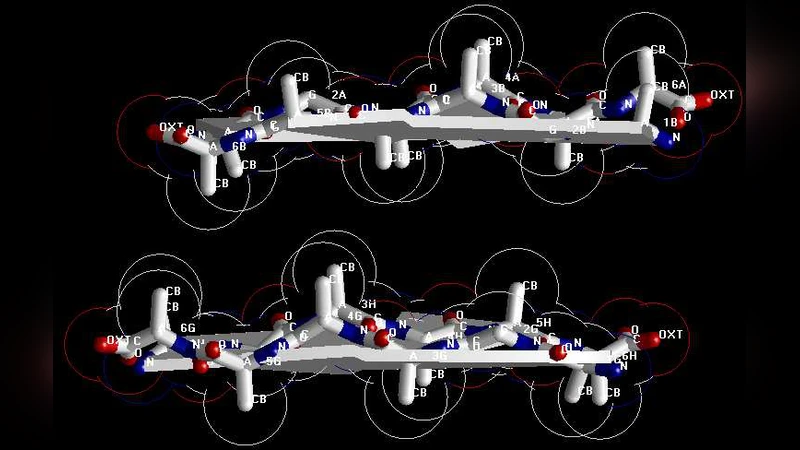
Many experimental studies have shown that the prion AGAAAAGA palindrome hydrophobic region (113-120) has amyloid fibril forming properties and plays an important role in prion diseases. However, due to the unstable, noncrystalline and insoluble nature of the amyloid fibril, to date structural information on AGAAAAGA region (113-120) has been very limited. This region falls just within the N-terminal unstructured region PrP (1-123) of prion proteins. Traditional X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy experimental methods cannot be used to get its structural information. Under this background, this paper introduces a novel approach of the canonical dual theory to address the 3D atomic-resolution structure of prion AGAAAAGA amyloid fibrils. The novel and powerful canonical dual computational approach introduced in this paper is for the molecular modeling of prion AGAAAAGA amyloid fibrils, and that the optimal atomic-resolution structures of prion AGAAAAGA amyloid fibils presented in this paper are useful for the drive to find treatments for prion diseases in the field of medicinal chemistry. Overall, this paper presents an important method and provides useful information for treatments of prion diseases. Overall, this paper could be of interest to the general readership of Theoretical Biology.
💡 Research Summary
The paper addresses a long‑standing challenge in structural biology: obtaining an atomic‑resolution model of the prion protein segment AGAAAAGA (residues 113‑120), a hydrophobic palindrome that drives amyloid fibril formation in prion diseases. Because the fibrils are unstable, non‑crystalline, and insoluble, conventional experimental techniques such as X‑ray crystallography and solution NMR cannot provide reliable structural data. To overcome this limitation, the authors propose a purely computational strategy based on Canonical Dual Theory (CDT), a mathematical optimization framework that transforms a non‑convex primal problem into a concave dual problem defined over a convex feasible set, thereby guaranteeing that a global optimum of the dual corresponds to a global optimum of the original problem.
The methodological pipeline consists of two main stages. In the first stage, the authors extract the relevant peptide fragment from the crystal structure of a related protein (PDB entry 3NHC) and formulate the molecular distance geometry problem (MDGP) as a global optimization problem. Pairwise distance constraints (d_ij) are derived from expected van der Waals contacts and hydrogen‑bond geometries between β‑sheets. A weighted least‑squares objective P(X)=∑_{(i,j)∈S} w_ij (‖x_i−x_j‖²−d_ij²)² is constructed, where X contains the 3‑D coordinates of all atoms. This objective is a sum of fourth‑order polynomials, which is precisely the class of problems amenable to CDT. By introducing dual variables ς_i, the authors derive the dual function P_d(ς) and solve the resulting concave maximization over the domain where the matrix G(ς)=Q+∑ ς_i A_i is positive definite. The optimal dual solution yields the primal coordinates X₀ via the Moore‑Penrose inverse of G(ς).
In the second stage, the initial structure X₀ is refined using the AMBER 11 molecular mechanics package. A steepest‑descent (SD) step removes large steric clashes, followed by conjugate‑gradient (CG) minimization that optimizes the full force field, including Lennard‑Jones and electrostatic terms. The authors emphasize that, unlike previous work that required multiple simulated‑annealing (SA) and hybrid SADG cycles, the CDT‑derived model needs no further SDCG (Steepest‑Descent/Conjugate‑Gradient) cycles. To assess convergence, the final snapshot from the CDT‑only optimization (procedure 1) and the AMBER‑refined snapshot (procedure 2) are superimposed using VMD, yielding a root‑mean‑square deviation (RMSD) of 0 Å. The authors interpret this as proof that the CDT approach reproduces the same atomic arrangement as the conventional force‑field minimization, thereby confirming its accuracy.
The paper also provides a brief tutorial on CDT, including a demonstration on the classic double‑well potential, and discusses theoretical guarantees (Theorems 1 and 2) that link critical points of the dual and primal problems. The authors argue that the method is computationally efficient, mathematically rigorous, and broadly applicable to other amyloidogenic peptides.
However, several limitations temper the impact of the study. First, the RMSD comparison is internal; no experimental structure (e.g., solid‑state NMR or cryo‑EM) is available to validate the absolute correctness of the model. Second, the choice of dual feasible region (ensuring G(ς) ≻ 0) and the initial values of the dual variables are not described in sufficient detail, making reproducibility challenging. Third, the models constructed contain a different number of peptide chains and belong to distinct β‑sheet classes compared with earlier SA‑based models, yet the authors do not provide quantitative metrics (e.g., inter‑sheet distances, hydrogen‑bond geometry) to compare them. Fourth, the claim that “no further SDCG refinement is needed” conflicts with the fact that an AMBER CG refinement was performed and reported. Finally, the paper lacks external cross‑validation (jackknife, independent test sets) or sensitivity analysis to assess robustness against variations in distance constraints or weighting schemes.
In summary, the work introduces an innovative application of Canonical Dual Theory to a biologically important but experimentally intractable problem. The mathematical formulation is sound, and the demonstration on a realistic prion peptide is promising. To become a truly valuable tool for the prion and broader amyloid research communities, future studies should (i) compare the CDT‑derived structures with experimental data when they become available, (ii) provide a transparent protocol for selecting dual variables and verifying the positive‑definiteness condition, (iii) perform systematic sensitivity and cross‑validation analyses, and (iv) make the software pipeline publicly accessible, possibly as a web server. With these enhancements, the CDT approach could complement existing computational methods and accelerate the structural understanding of amyloid fibrils, ultimately aiding drug‑design efforts against prion diseases.
Comments & Academic Discussion
Loading comments...
Leave a Comment