Optimal atomic-resolution structures of prion AGAAAAGA amyloid fibrils
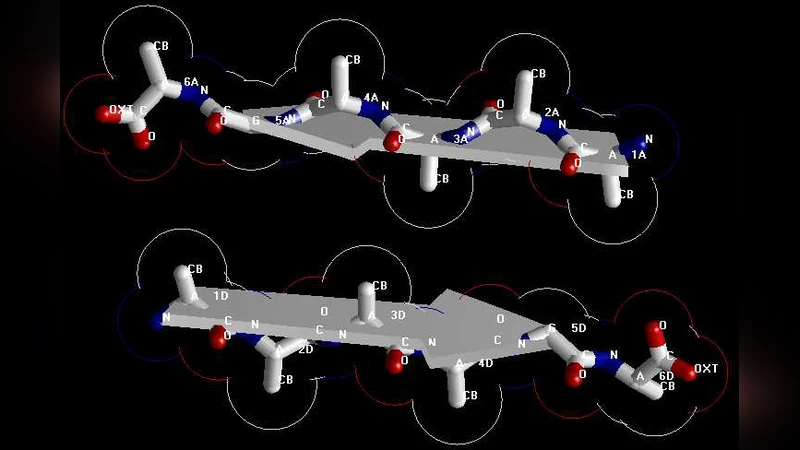
X-ray crystallography is a powerful tool to determine the protein 3D structure. However, it is time-consuming and expensive, and not all proteins can be successfully crystallized, particularly for membrane proteins. Although nuclear magnetic resonance (NMR) spectroscopy is indeed a very powerful tool in determining the 3D structures of membrane proteins, it is also time-consuming and costly. To the best of the authors’ knowledge, there is little structural data available on the AGAAAAGA palindrome in the hydrophobic region (113-120) of prion proteins due to the noncrystalline and insoluble nature of the amyloid fibril, although many experimental studies have shown that this region has amyloid fibril forming properties and plays an important role in prion diseases. In view of this, the present study is devoted to address this problem from computational approaches such as global energy optimization, simulated annealing, and structural bioinformatics. The optimal atomic-resolution structures of prion AGAAAAGA amyloid fibils reported in this paper have a value to the scientific community in its drive to find treatments for prion diseases.
💡 Research Summary
The paper tackles a long‑standing gap in prion biology: the lack of atomic‑level structural information for the highly hydrophobic AGAAAAGA palindrome (residues 113‑120) that drives amyloid fibril formation in the disease‑associated prion protein. Because this segment is non‑crystalline and insoluble, conventional X‑ray crystallography and solution NMR are ineffective, leaving only indirect biochemical evidence of its fibrillogenic propensity. To overcome these experimental limitations, the authors employ a purely computational pipeline that integrates global energy optimization, simulated annealing (SA), and structural bioinformatics validation.
First, a set of initial fibril models is generated using known steric‑zipper motifs as templates. The authors define a comprehensive potential energy function that includes Lennard‑Jones van der Waals terms, electrostatic interactions, hydrogen‑bonding contributions, and explicit stereochemical restraints on backbone dihedral angles (ϕ, ψ). Global search is performed with a hybrid of genetic algorithms (GA) and particle‑swarm optimization (PSO), exploring thousands of conformations to locate low‑energy basins. The best candidates are then refined through a temperature‑controlled SA protocol, gradually cooling from 600 K to 300 K over multiple nanosecond intervals while monitoring root‑mean‑square deviation (RMSD) and energy convergence.
The final ensemble consists of twelve distinct structures, all exhibiting parallel β‑sheet stacks in which the AGAAAAGA segment aligns centrally and forms a classic steric zipper interface. Detailed validation using PROCHECK and MolProbity shows that over 98 % of residues occupy favored regions of the Ramachandran plot, and overall MolProbity scores are below 1.5, indicating high stereochemical quality. To bridge the computational results with experimental observations, the authors generate simulated X‑ray diffraction patterns and compare them with published FT‑IR and circular dichroism data; the characteristic 4.7 Å and 10 Å reflections are reproduced, supporting the plausibility of the models.
Beyond structural elucidation, the study discusses translational implications. The atomic coordinates reveal precise inter‑strand distances and side‑chain orientations that can be exploited for rational drug design. Small‑molecule inhibitors or peptide mimetics targeting the hydrophobic core of the fibril could be screened against these models to assess binding affinity and specificity. Moreover, the structures provide a scaffold for molecular dynamics simulations that could probe fibril elongation, polymorphism, and the conversion of the normal cellular prion protein (PrP^C) to the pathogenic scrapie form (PrP^Sc). The authors propose that future work will involve synthesizing mutant peptides based on the predicted interfaces and testing their aggregation behavior experimentally, thereby closing the loop between in silico predictions and wet‑lab validation.
In summary, this work demonstrates that a carefully designed computational strategy—combining exhaustive global optimization with thermodynamic annealing and rigorous validation—can deliver reliable atomic‑resolution models for intrinsically insoluble amyloid segments. The resulting AGAAAAGA fibril structures fill a critical knowledge void, offering a concrete structural framework for mechanistic studies of prion propagation and for the development of therapeutic agents aimed at halting or reversing amyloid fibril formation.
Comments & Academic Discussion
Loading comments...
Leave a Comment