MCUIUC -- A New Framework for Metagenomic Read Compression
Metagenomics is an emerging field of molecular biology concerned with analyzing the genomes of environmental samples comprising many different diverse organisms. Given the nature of metagenomic data, one usually has to sequence the genomic material of all organisms in a batch, leading to a mix of reads coming from different DNA sequences. In deep high-throughput sequencing experiments, the volume of the raw reads is extremely high, frequently exceeding 600 Gb. With an ever increasing demand for storing such reads for future studies, the issue of efficient metagenomic compression becomes of paramount importance. We present the first known approach to metagenome read compression, termed MCUIUC (Metagenomic Compression at UIUC). The gist of the proposed algorithm is to perform classification of reads based on unique organism identifiers, followed by reference-based alignment of reads for individually identified organisms, and metagenomic assembly of unclassified reads. Once assembly and classification are completed, lossless reference based compression is performed via positional encoding. We evaluate the performance of the algorithm on moderate sized synthetic metagenomic samples involving 15 randomly selected organisms and describe future directions for improving the proposed compression method.
💡 Research Summary
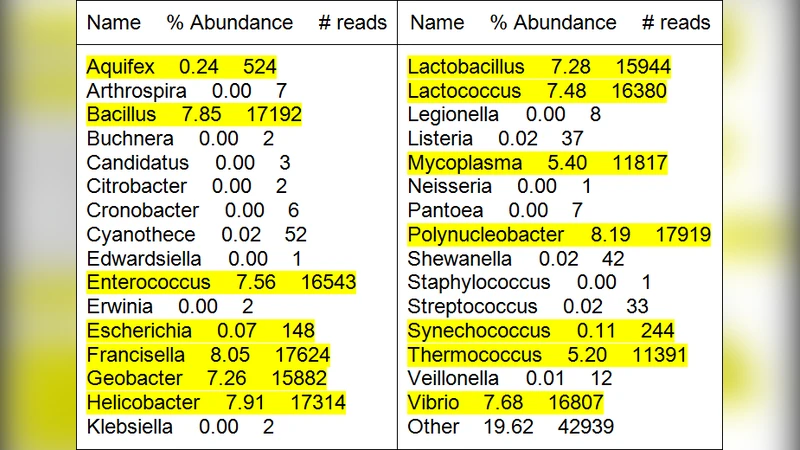
The paper introduces MCUIUC (Metagenomic Compression at UIUC), the first systematic framework designed to achieve lossless compression of large‑scale metagenomic sequencing reads. Metagenomic experiments generate massive volumes of short reads (often >600 GB) from a mixture of many microbial genomes, making storage and transmission a critical bottleneck. Existing compression methods focus on single‑genome data and cannot be directly applied because the source organism of each read is unknown. MCUIUC addresses this by first performing a coarse taxonomic classification of the reads at the genus level using the Metaphyler tool, which scans for unique marker sequences longer than 20 bp. Metaphyler outputs a list of candidate genera together with estimated abundances; a user‑defined abundance threshold (e.g., 0.1 %) filters out low‑confidence candidates.
Once a set of genera is selected, the pipeline proceeds to partition the read set. All reference genomes belonging to the identified genera are retrieved from public databases, and Bowtie2 is used to align the reads against each genome in a high‑throughput manner. For each genus, the species that attracts the largest number of aligned reads is designated as the “representative” genome. All reads assigned to that genus are then aligned to this representative, and their alignment positions and mismatches are encoded using a Golomb‑code based positional encoding scheme originally described for reference‑based genome compression. Reads that fail to align (typically 20–25 % of the total) are treated separately: they are assembled into contigs using a metagenomic assembler such as IDBA‑UD, and the resulting contigs are queried against NCBI databases (via BLAST) to infer their likely origin. If new genera are discovered among the contigs, the classification‑alignment‑compression cycle is repeated recursively.
The compression stage converts the per‑genus SAM files into sorted BAM files with SAMtools, then applies the CRAM toolkit to obtain reference‑based compressed files (CRAM) that store only the positional differences relative to the representative genome. Unaligned reads, the list of representative genomes, Metaphyler’s taxonomic report, and any auxiliary reference sequences are compressed with standard tools (e.g., bzip2) and bundled into a tar archive for distribution. Decompression is straightforward: the CRAM files are decoded with the stored references, and the original FASTA reads are perfectly reconstructed.
To evaluate the method, the authors generated a synthetic metagenomic dataset consisting of 15 bacterial species randomly drawn from the NCBI microbial genome collection. Each genome was simulated at 100× coverage using Illumina‑style paired reads, producing a 5 GiB FASTA file. Metaphyler identified 31 genera; after applying a 0.1 % abundance filter, the 15 true genera were retained. Bowtie2 alignment succeeded for roughly 75 % of the reads, while the remaining 25 % were assembled into contigs with IDBA‑UD. Compression results showed an average 6‑fold reduction per genus when using CRAM, and an overall archive size about 5.8 times smaller than the original FASTA.
The authors acknowledge several limitations. The accuracy of the initial Metaphyler classification directly influences the choice of representative genomes; misclassifications can degrade compression efficiency. The assembly step remains computationally intensive and may become a bottleneck for real‑world samples containing hundreds of species. Future work is proposed in four main directions: (1) employing deep‑learning‑based marker detection to improve taxonomic resolution, (2) implementing dynamic representative genome replacement to adapt to classification errors, (3) exploring multi‑reference compression that leverages assembled contigs as additional references, and (4) scaling the pipeline through distributed computing frameworks and cloud resources.
In summary, MCUIUC demonstrates that a hierarchical approach—first reducing the taxonomic complexity of a metagenomic sample, then applying proven single‑genome compression techniques—can achieve substantial lossless compression of metagenomic reads. This framework offers a practical solution for the growing storage demands of metagenomic projects and provides a foundation for further algorithmic enhancements.
Comments & Academic Discussion
Loading comments...
Leave a Comment