Extending Hirshfeld-I to bulk and periodic materials
In this work, a method is described to extend the iterative Hirshfeld-I method, generally used for molecules, to periodic systems. The implementation makes use of precalculated pseudo-potential based charge density distributions, and it is shown that high quality results are obtained for both molecules and solids, such as ceria, diamond, and graphite. The use of such grids makes the implementation independent of the solid state or quantum chemical code used for studying the system. The extension described here allows for easy calculation of atomic charges and charge transfer in periodic and bulk systems.
💡 Research Summary
The paper presents a practical extension of the iterative Hirshfeld‑I (Hirshfeld‑I) charge‑analysis method, originally designed for isolated molecules, to periodic bulk and surface systems. The authors address two fundamental challenges that arise when moving from finite molecular clusters to infinite crystals: (i) the need to truncate the formally infinite sum over all atoms in the crystal, and (ii) the computational cost of repeatedly evaluating the electron density from wave‑functions.
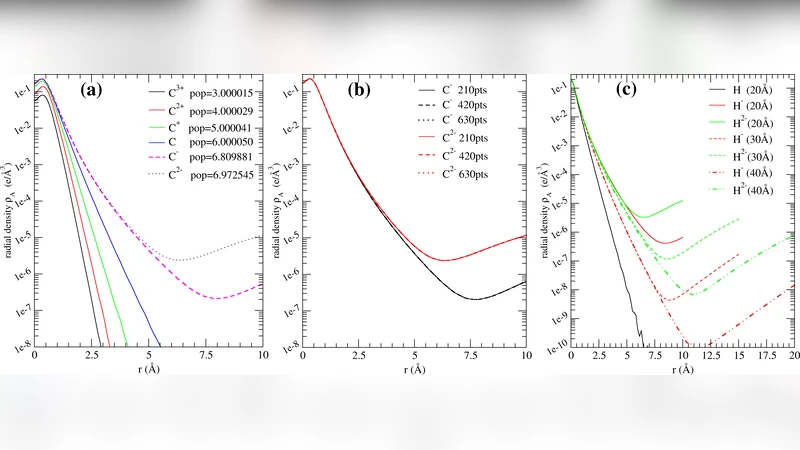
To solve (i), they introduce a “Sphere of Influence” (SoI) concept. Because atomic charge densities decay exponentially with distance, contributions from atoms beyond a certain radius become negligible. By defining a finite SoI radius (typically ~6 Å) around each atom, the infinite sum in the Hirshfeld‑I weight equations can be safely truncated without compromising accuracy. This truncation is justified by systematic tests showing that enlarging the SoI beyond this value does not change the resulting atomic charges beyond a few micro‑electrons.
For (ii), the authors exploit the fact that most plane‑wave DFT codes (e.g., VASP, Quantum ESPRESSO) can output the total electron density on a three‑dimensional grid. They pre‑compute and store two types of grids: (a) a linear grid for the whole periodic system (V_sys) and (b) a linear grid for each isolated reference atom or ion (V_atom). During the Hirshfeld‑I iterations, the algorithm interpolates these grids rather than recomputing densities from the underlying wave‑functions. This grid‑based approach makes the implementation completely independent of the electronic‑structure code used to generate the densities, while dramatically reducing computational overhead.
The integration of atomic populations is performed using Becke’s multicenter scheme. Atom‑centered spherical grids (S_atom for reference atoms and S_sys for the target system) are constructed from a logarithmic radial grid and a Lebedev‑Laikov angular grid. A smooth weight function h_A(r) ensures that overlapping regions are counted only once, eliminating double‑counting errors inherent in naive partitioning.
A subtle issue arises from the use of PAW pseudopotentials, which freeze core electrons. Consequently, the stored atomic charge densities contain only valence electrons, and near the nucleus the radial density can exhibit non‑monotonic behavior (even negative values). The authors argue that because both the system density and the reference atomic densities are generated with the same PAW potentials, any systematic error cancels out in the Hirshfeld‑I partitioning.
Convergence is declared when the maximum change in any atomic charge between successive iterations falls below 5 × 10⁻⁴ e. Tests on a linear grid resolution of 0.05 Å show that charges converge within a few tens of micro‑electrons, confirming the robustness of the interpolation scheme.
The methodology is validated on several benchmark systems. For the CO molecule, Hirshfeld‑I charges obtained from the grid‑based periodic implementation agree with those from traditional Gaussian‑03 calculations (using B3LYP/6‑311G** and a dense Becke integration grid). For bulk ceria (CeO₂), diamond, and graphite, the computed atomic charges reflect chemically sensible oxidation states (e.g., Ce ≈ +3.9, O ≈ –1.9 in CeO₂) and reproduce known trends such as the near‑zero charge transfer in pristine graphite. These results demonstrate that the method works equally well for isolated molecules and extended solids.
Compared with existing implementations (e.g., Cut3D in ABINIT, DDEC in the Manz‑Sholl code, or subsystem‑based Hirshfeld‑I approaches), the present scheme offers three distinct advantages: (1) complete code‑independence—any DFT package that can output a charge‑density grid suffices; (2) simplicity—no need for specialized post‑processing tools beyond standard grid interpolation; and (3) physical fidelity—by retaining the smooth, weighted nature of Hirshfeld‑I, the method avoids the abrupt partitioning of Bader analysis while still providing chemically intuitive atomic charges.
In conclusion, the authors deliver a robust, efficient, and broadly applicable extension of Hirshfeld‑I to periodic materials. The approach enables routine, high‑quality atomic charge analysis for bulk crystals, surfaces, and low‑dimensional materials, opening the door to systematic studies of charge transfer in defects, dopants, adsorption phenomena, and complex metal‑organic frameworks. Future work may focus on automating SoI selection, integrating with machine‑learning force fields, and applying the method to strongly correlated oxides where charge localization plays a pivotal role.
Comments & Academic Discussion
Loading comments...
Leave a Comment