Computational Assignment of Chemical Shifts for Protein Residues
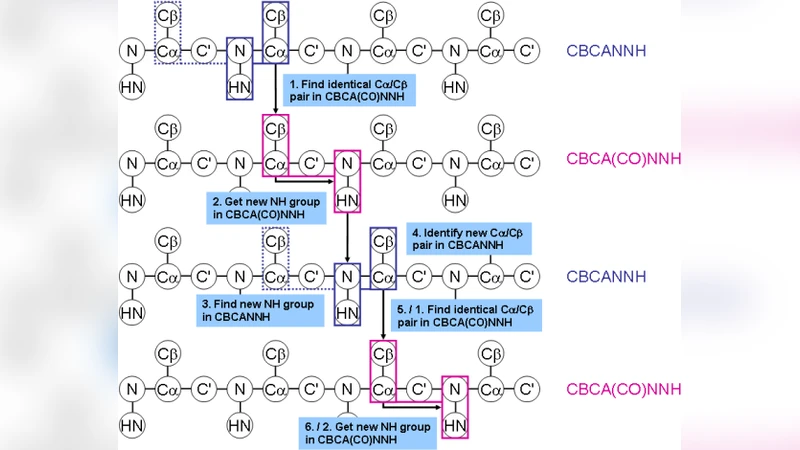
Fast and accurate protein structure prediction is one of the major challenges in structural biology, biotechnology and molecular biomedicine. These fields require 3D protein structures for rational design of proteins with improved or novel properties. X-ray crystallography is the most common approach even with its low success rate, but lately NMR based approaches have gained popularity. The general approach involves a set of distance restraints used to guide a structure prediction, but simple NMR triple-resonance experiments often provide enough structural information to predict the structure of small proteins. Previous protein folding simulations that have utilised experimental data have weighted the experimental data and physical force field terms more or less arbitrarily, and the method is thus not generally applicable to new proteins. Furthermore a complete and near error-free assignment of chemical shifts obtained by the NMR experiments is needed, due to the static, or deterministic, assignment. In this thesis I present Chemshift, a module for handling chemical shift assignments, implemented in the protein structure determination program Phaistos. This module treats both the assignment of experimental data, as well as the weighing compared to physical terms, in a probabilistic framework where no data is discarded. Provided a partial assignment of NMR peaks, the module is able to improve the assignment with the intension to utilise this in the protein folding with little bias.
💡 Research Summary
**
The thesis presents Chemshift, a novel module for handling NMR chemical‑shift assignments within the protein structure prediction framework Phaistos. The author identifies three major bottlenecks in current NMR‑based structure determination: (1) the labor‑intensive and error‑prone manual assignment of chemical‑shift peaks, (2) the arbitrary weighting of experimental data versus physical force‑field terms in hybrid energy functions, and (3) the loss of valuable data when deterministic assignments fail to converge during folding simulations.
Chemshift addresses these issues by embedding the assignment problem in a fully probabilistic Bayesian framework. Measured chemical shifts are modeled as Gaussian random variables with mean μ (the true shift) and standard deviation σ that captures both experimental noise and prediction uncertainty. The joint probability of two independent measurements is derived analytically, yielding a simple expression proportional to exp
Comments & Academic Discussion
Loading comments...
Leave a Comment