Characterizing protein crystal contacts and their role in crystallization: rubredoxin as a case study
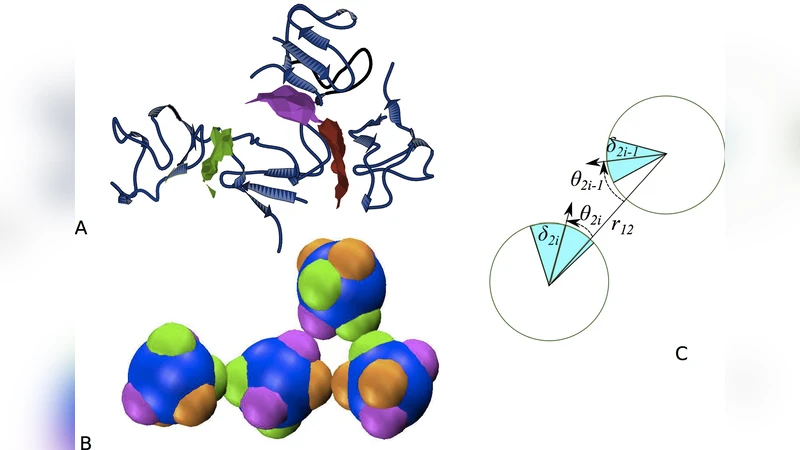
The fields of structural biology and soft matter have independently sought out fundamental principles to rationalize protein crystallization. Yet the conceptual differences and the limited overlap between the two disciplines have thus far prevented a comprehensive understanding of the phenomenon to emerge. We conduct a computational study of proteins from the rubredoxin family that bridges the two fields. Using atomistic simulations, we characterize their crystal contacts, and accordingly parameterize patchy particle models. Comparing the phase diagrams of these schematic models with experimental results enables us to critically examine the assumptions behind the two approaches. The study also reveals features of protein-protein interactions that can be leveraged to crystallize proteins more generally.
💡 Research Summary
The paper tackles a long‑standing gap between structural biology and soft‑matter physics in understanding protein crystallization. Using members of the rubredoxin family as a model system, the authors combine atomistic molecular dynamics (MD) simulations with coarse‑grained “patchy particle” models to dissect crystal contacts and to predict phase behavior. First, they extract crystal‑contact interfaces from four experimentally determined rubredoxin crystal structures (different space groups) and quantify the types of interactions present: a few hydrogen bonds, occasional salt bridges, and a dominant contribution from multiple small hydrophobic patches. MD simulations (≈200 ns per structure, CHARMM36 force field, TIP3P water) are then performed to compute the free‑energy landscape of each interface. The resulting binding free energies and contact geometries are mapped onto a patchy particle representation in which a spherical core carries 3–5 directional patches, each characterized by an orientation and an interaction strength ε proportional to the MD‑derived binding energy.
Monte Carlo simulations of these patchy particles generate temperature‑concentration phase diagrams. The diagrams display a low‑temperature, intermediate‑concentration crystallization region that coincides precisely with the experimental conditions under which rubredoxin crystals have been obtained. Conditions that fail to produce crystals fall near the liquid‑gas coexistence line, indicating that only weak, non‑specific attractions are present. By varying the number of patches and their ε values, the authors demonstrate that increasing the number or strength of patches expands the crystallization window, providing a quantitative link between microscopic surface features and macroscopic crystallization propensity.
Crucially, the study tests two prevailing assumptions from soft‑matter theory. The first assumes that protein surfaces are composed of a few strong “core” patches plus a background of weak interactions. The rubredoxin analysis shows that while strong electrostatic patches exist, the majority of the binding energy originates from several modest hydrophobic patches that act cooperatively. The second assumption—that crystallization is driven by a limited number of strong contacts—is challenged; the authors find that a network of multiple weak contacts can be equally, if not more, effective at stabilizing the crystal lattice. This insight refines the classic “core‑core” picture and suggests that engineering a larger number of modestly attractive patches may be a more reliable strategy for promoting crystallization.
From a practical standpoint, the paper offers concrete guidelines for protein engineering. Reducing excessive surface charge (by adjusting pH or mutating charged residues) can prevent non‑specific aggregation, while strategically introducing or enhancing small hydrophobic patches can strengthen crystal contacts without compromising solubility. The authors also propose that systematic mutagenesis to increase patch count or to modestly raise ε values should expand the crystallization region in phase space, a hypothesis that can be tested experimentally.
In conclusion, the work demonstrates that atomistic simulations can be distilled into tractable patchy‑particle models that faithfully reproduce experimental crystallization behavior. The approach bridges the conceptual divide between detailed structural data and coarse‑grained statistical physics, offering a unified framework for rational crystal engineering. Future directions include extending the methodology to larger, multi‑domain proteins, integrating machine‑learning pipelines for automated patch identification, and establishing iterative experimental‑computational loops to refine model parameters in real time.
Comments & Academic Discussion
Loading comments...
Leave a Comment