Improving transcriptome assembly through error correction of high-throughput sequence reads
The study of functional genomics–particularly in non-model organisms has been dramatically improved over the last few years by use of transcriptomes and RNAseq. While these studies are potentially extremely powerful, a computationally intensive procedure–the de novo construction of a reference transcriptome must be completed as a prerequisite to further analyses. The accurate reference is critically important as all downstream steps, including estimating transcript abundance are critically dependent on the construction of an accurate reference. Though a substantial amount of research has been done on assembly, only recently have the pre-assembly procedures been studied in detail. Specifically, several stand-alone error correction modules have been reported on, and while they have shown to be effective in reducing errors at the level of sequencing reads, how error correction impacts assembly accuracy is largely unknown. Here, we show via use of a simulated dataset, that applying error correction to sequencing reads has significant positive effects on assembly accuracy, by reducing assembly error by nearly 50%, and therefore should be applied to all datasets.
💡 Research Summary
The manuscript investigates the impact of pre‑assembly error correction (EC) on de novo transcriptome assembly quality, focusing on Illumina RNA‑seq data. The authors note that Illumina reads exhibit a non‑random error profile, with substitution errors increasing toward the 3′ end and overall error rates ranging from 1 % to 3 %. Such errors inflate the complexity of de Bruijn graphs used by most assemblers, leading to higher memory consumption, longer runtimes, and, critically, nucleotide‑level inaccuracies in the resulting transcriptome. While many error‑correction tools have been developed for genomic data, their effect on RNA‑seq assemblies has not been systematically evaluated.
To address this gap, the study uses two datasets. The primary dataset is a simulated set of 30 million paired‑end 100‑nt reads generated with Flux Simulator, based on the Mus musculus transcriptome, achieving an average coverage of ~70× and incorporating a realistic Illumina error model. The second dataset is an empirical set of 50 million 76‑nt paired‑end reads from mouse mRNA, from which 30 million reads are randomly selected for analysis. Both datasets are processed with four widely used error‑correction packages: Reptile, SGA, AllPaths‑LG, and Seecer (the latter being RNA‑seq‑specific). The authors report the number of corrected nucleotides, runtime, and the proportion of reads affected for each tool (Table 1).
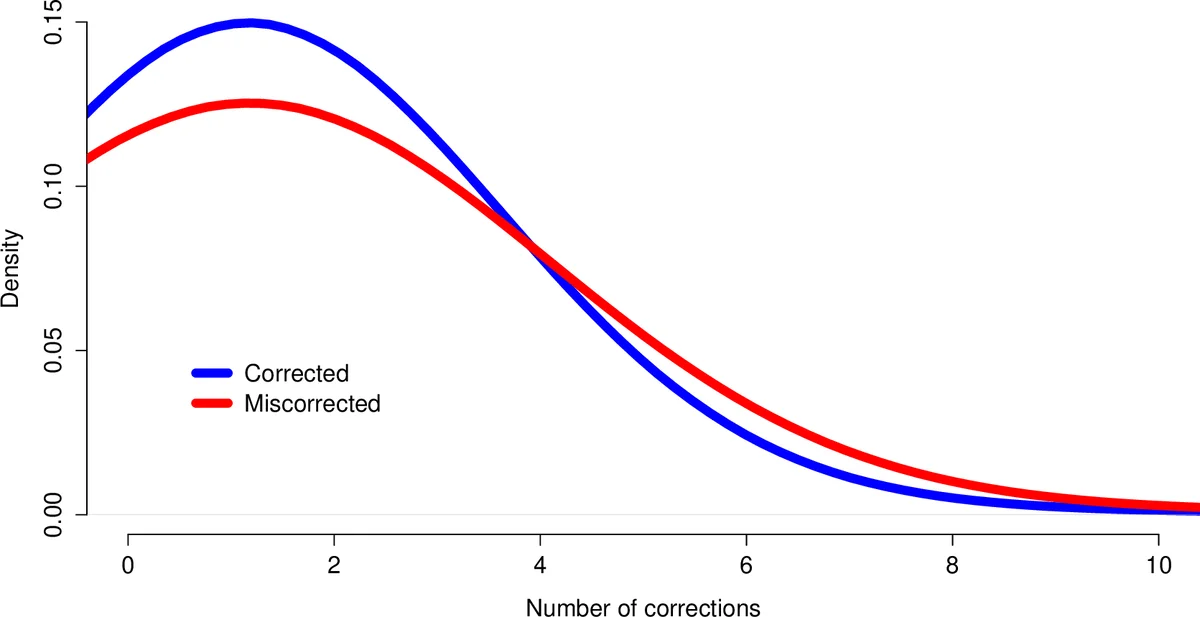
After correction, each read set is assembled with Trinity using identical parameters. Assembly quality is assessed through multiple metrics: total assembly size, N50, the number of “high‑confidence” contigs (≥99 % identity to reference and covering ≥90 % of the reference length), average per‑contig mismatches, and the proportion of reads that map back to the assembly.
Key findings from the simulated data:
- The raw‑read assembly contains 38 459 high‑confidence contigs with an average of 1.40 mismatches per contig (≈1.40 % error rate).
- Reptile correction reduces the average mismatches to 1.23 per contig (≈12 % reduction) and yields the largest high‑confidence set (38 670 contigs). It corrects ~7.8 million nucleotides (≈0.16 % of the total bases) but requires multiple runs and >12 hours of manual tuning.
- SGA is the fastest (≈38 minutes), correcting ~19.8 million nucleotides and achieving a modest 4 % error reduction.
- AllPaths‑LG aggressively corrects ~140 million nucleotides (≈2.7 % error reduction) within ~8 hours, but its high‑confidence set is smaller than that of Reptile.
- Seecer, despite being designed for RNA‑seq, changes ~8.8 million nucleotides yet does not improve the mismatch rate relative to the raw assembly.
For the empirical dataset, trends are consistent: Reptile again yields the greatest reduction in nucleotide errors (≈10 % decrease) and the largest high‑confidence contig set (21 580 contigs). SGA also performs well, reducing errors by >9 %. AllPaths‑LG and Seecer show limited gains. Mapping rates improve modestly across all corrected datasets, rising from 92.44 % (raw) to a maximum of 94.89 % (SGA‑corrected).
Importantly, global assembly statistics such as total size, N50, and contig length distributions remain largely unchanged across correction methods, indicating that error correction primarily refines base‑level accuracy rather than gross contiguity. Nevertheless, the reduction of nucleotide‑level errors is crucial for downstream analyses that depend on precise sequence information, such as SNP discovery, allele‑specific expression, and accurate quantification of transcript abundance.
The authors conclude that error correction should become a routine preprocessing step for RNA‑seq projects, especially when de novo assembly is required. Among the tools evaluated, Reptile provides the most consistent improvement in both simulated and real data, albeit at the cost of computational time and manual parameter optimization. SGA offers a rapid alternative with respectable gains, while AllPaths‑LG and Seecer may be less suitable for transcriptome assemblies under the conditions tested. The study also highlights the need for more automated, transcriptome‑aware error‑correction algorithms that can balance correction aggressiveness with preservation of true biological variation.
Overall, this work provides a comprehensive, data‑driven assessment of how pre‑assembly error correction influences transcriptome assembly quality, offering practical guidance for researchers and underscoring the importance of addressing sequencing errors before downstream functional genomics analyses.
Comments & Academic Discussion
Loading comments...
Leave a Comment