Application of three graph Laplacian based semi-supervised learning methods to protein function prediction problem
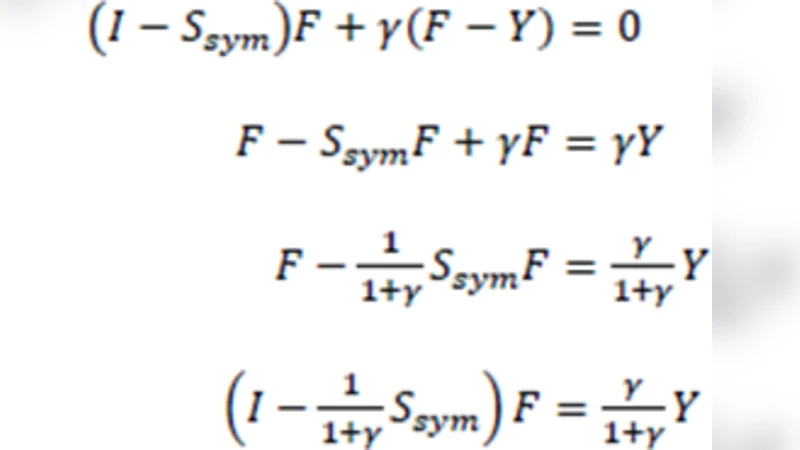
Protein function prediction is the important problem in modern biology. In this paper, the un-normalized, symmetric normalized, and random walk graph Laplacian based semi-supervised learning methods will be applied to the integrated network combined from multiple networks to predict the functions of all yeast proteins in these multiple networks. These multiple networks are network created from Pfam domain structure, co-participation in a protein complex, protein-protein interaction network, genetic interaction network, and network created from cell cycle gene expression measurements. Multiple networks are combined with fixed weights instead of using convex optimization to determine the combination weights due to high time complexity of convex optimization method. This simple combination method will not affect the accuracy performance measures of the three semi-supervised learning methods. Experiment results show that the un-normalized and symmetric normalized graph Laplacian based methods perform slightly better than random walk graph Laplacian based method for integrated network. Moreover, the accuracy performance measures of these three semi-supervised learning methods for integrated network are much better than the best accuracy performance measures of these three methods for the individual network.
💡 Research Summary
The paper investigates the use of three graph‑Laplacian‑based semi‑supervised learning (SSL) algorithms—unnormalized, symmetric normalized, and random‑walk Laplacians—for predicting the functions of Saccharomyces cerevisiae proteins. The authors first construct five heterogeneous biological networks that capture complementary evidence about protein relationships: (1) a Pfam domain‑based similarity network, (2) a protein‑complex co‑participation network, (3) a protein‑protein interaction (PPI) network, (4) a genetic interaction network, and (5) a cell‑cycle gene‑expression correlation network. Each network is represented by an adjacency matrix (A_i) and a degree matrix (D_i).
Instead of solving a computationally intensive convex‑optimization problem to learn optimal combination weights, the authors simply assign equal weights to all five networks and form an integrated adjacency matrix (A = \frac{1}{5}\sum_{i=1}^{5} A_i). This “fixed‑weight” strategy dramatically reduces runtime and memory consumption while preserving predictive performance, as demonstrated experimentally.
For SSL, a small subset of proteins (approximately 10 % of the total) with known Gene Ontology (GO) annotations is used as the seed label matrix (Y_0). The three Laplacians are defined as follows:
- Unnormalized Laplacian (L = D - A) (transition matrix (S = A)).
- Symmetric normalized Laplacian (L_{\text{sym}} = I - D^{-1/2} A D^{-1/2}) (transition matrix (S = D^{-1/2} A D^{-1/2})).
- Random‑walk Laplacian (L_{\text{rw}} = I - D^{-1} A) (transition matrix (S = D^{-1} A)).
Label propagation follows the classic iterative rule
\
Comments & Academic Discussion
Loading comments...
Leave a Comment