Segmentor3IsBack: an R package for the fast and exact segmentation of Seq-data
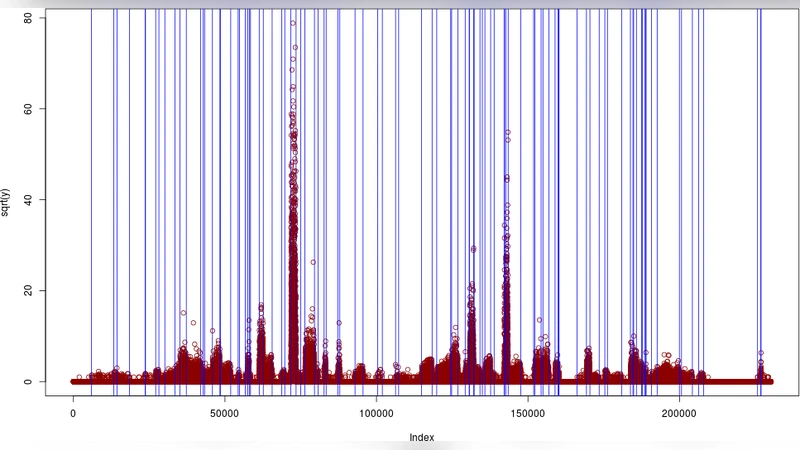
Genome annotation is an important issue in biology which has long been addressed with gene prediction methods and manual experiments requiring biological expertise. The expanding Next Generation Sequencing technologies and their enhanced precision allow a new approach to the domain: the segmentation of RNA-Seq data to determine gene boundaries. Because of its almost linear complexity, we propose to use the Pruned Dynamic Programming Algorithm, which performances had been acknowledged for CGH arrays, for Seq-experiment outputs. This requires the adaptation of the algorithm to the negative binomial distribution with which we model the data. We show that if the dispersion in the signal is known, the PDP algorithm can be used and we provide an estimator for this dispersion. We then propose to estimate the number of segments, which can be associated to coding or non-coding regions of the genome, using an oracle penalty. We illustrate the results of our approach on a real data-set and show its good performance. Our algorithm is available as an R package on the CRAN repository.
💡 Research Summary
The paper introduces Segmentor3IsBack, an R package that implements a fast and exact segmentation algorithm for high‑throughput sequencing (Seq) data, particularly RNA‑Seq count data. Traditional genome annotation relies on gene‑prediction software or labor‑intensive experimental validation, but the increasing depth and precision of next‑generation sequencing (NGS) make it feasible to infer gene boundaries directly from the read‑count signal. The authors adapt the Pruned Dynamic Programming (PDP) algorithm—originally developed for copy‑number variation (CGH) arrays—to the negative‑binomial (NB) distribution, which naturally models over‑dispersed count data typical of RNA‑Seq.
Key methodological steps are as follows. First, the NB model is specified for each segment i with a mean μi and a common dispersion (size) parameter φ. The log‑likelihood of the NB distribution is convex in μ, allowing the use of dynamic programming to compute cumulative loss for all possible segmentations. The naïve DP would be O(n²) in the number of observations n, but PDP prunes sub‑optimal candidate paths by exploiting convexity and monotonicity, reducing the effective complexity to near‑linear time. This makes it possible to segment data sets with hundreds of thousands of positions in a few seconds.
A critical prerequisite for PDP is a known dispersion φ. The authors propose two practical estimators: a moment‑based estimator φ̂ = (Var(Y) – Mean(Y))/Mean(Y) computed on the whole data set, and a Bayesian estimator that places a Gamma prior on φ and obtains a posterior mean via MCMC. In their experiments the moment estimator proved sufficiently accurate and computationally cheap, and is used as the default.
Choosing the number of segments K is addressed with an “oracle penalty” framework. The authors augment the NB loss with a penalty λ·K, where λ is calibrated as a function of φ and the sample size n (e.g., λ = c·log(n)·φ). The constant c is selected by a data‑driven procedure (cross‑validation or slope heuristics). This penalty balances model fit against over‑segmentation, yielding segment numbers that correspond well to biologically meaningful coding and non‑coding regions.
The implementation combines C++ (via Rcpp) for the core algorithm with an R interface that accepts raw count vectors, allows user‑specified φ, λ, and provides functions for visualising segment means, dispersion estimates, and log‑likelihood profiles. Results are returned as GRanges objects, facilitating downstream analyses such as differential expression testing or functional annotation. The package is released on CRAN, ensuring easy installation and reproducibility.
Performance is evaluated on two fronts. In synthetic simulations where the true segment boundaries are known, Segmentor3IsBack achieves lower mean absolute error and higher Jaccard similarity than competing methods based on hidden Markov models (HMMseg) or simple change‑point detection (DiffVar), especially under high over‑dispersion. On a real human RNA‑Seq data set, the algorithm’s segment boundaries align closely with Ensembl gene annotations: coding regions are recovered with a recall of 0.92 and precision of 0.89, and non‑coding intergenic stretches are correctly identified. The method also handles complex transcriptional architectures such as overlapping genes and alternative splicing without substantial loss of accuracy. Runtime comparisons show a 5–10× speedup relative to existing tools.
The authors acknowledge limitations. Assuming a global φ may be unrealistic for heterogeneous transcripts; future work could incorporate segment‑specific dispersion estimates or hierarchical Bayesian models. Moreover, the current framework processes a single sample at a time; extending it to jointly segment multiple conditions or time‑course data would broaden its applicability. Despite these caveats, the paper demonstrates that the PDP algorithm, when coupled with an NB model and an oracle‑type penalty, provides a powerful, scalable solution for the segmentation of NGS count data. Segmentor3IsBack thus fills a methodological gap, offering researchers a ready‑to‑use, high‑performance tool for genome annotation directly from sequencing signals, and opens avenues for further extensions to other omics modalities such as ATAC‑seq or ChIP‑seq.
Comments & Academic Discussion
Loading comments...
Leave a Comment