Towards Kinetic Modeling of Global Metabolic Networks with Incomplete Experimental Input on Kinetic Parameters
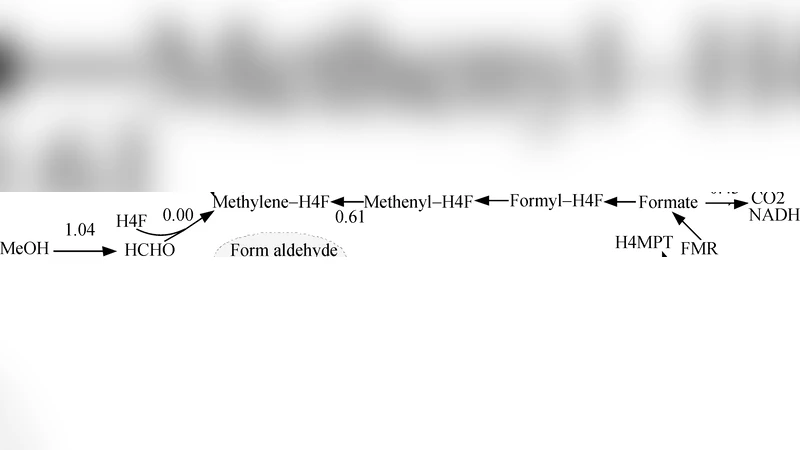
This is the first report, to our knowledge, on a systematic method for constructing a large scale kinetic metabolic model with incomplete information on kinetic parametersr, and its initial application to the modeling of central metabolism of Methylobacterium extorquens AM1, a methylotrophic and environmental important bacterium, with all necessary constraints. Through a systematic and consistent procedure of finding a set of parameters in the physiological range we overcome an outstanding difficulty in large scale kinetic modeling: the requirement for a massive number of enzymatic reaction parameters. We are able to construct the kinetic model based on general biological considerations and incomplete experimental kinetic parameters. The success of our approach with incompletely input information is guaranteed by two known principles in biology, the robustness of the system and the cooperation among its various parts. (Will be pleased to be informed on other methodologies dealing with same type of problems: aoping@u.washington.edu)
💡 Research Summary
The paper addresses one of the most persistent challenges in systems biology: constructing large‑scale kinetic models of metabolism when the majority of enzyme kinetic parameters (Km, Vmax, inhibition constants, etc.) are unknown or only partially characterized. Traditional approaches either limit themselves to small pathways where exhaustive kinetic data can be gathered, or they resort to steady‑state flux balance analysis (FBA) that ignores enzyme dynamics altogether. The authors propose a systematic, reproducible workflow that leverages two well‑established biological principles—system robustness and cooperative interaction among pathway components—to infer physiologically plausible kinetic parameter sets despite incomplete experimental input.
The workflow consists of four sequential stages. First, known kinetic values are collected from literature and databases such as BRENDA and SABIO‑RK; missing parameters are sampled from broad, biologically reasonable ranges (log‑uniform distributions spanning micromolar to millimolar concentrations and realistic Vmax values). Second, each sampled parameter set is subjected to thermodynamic consistency checks: the standard Gibbs free energy change (ΔG°′) for every reaction is calculated, adjusted for intracellular pH, ionic strength, and metabolite concentrations, and reactions with positive ΔG are discarded as thermodynamically infeasible. Third, the remaining candidates are filtered through physiological constraints derived from measured intracellular metabolite levels, substrate uptake rates, and overall flux distributions obtained from experimental chemostat cultures of Methylobacterium extorquens AM1. This step enforces the “robustness” principle—only those parameter combinations that produce fluxes within experimentally observed bounds, and that remain relatively stable under modest perturbations, are retained. Fourth, the authors incorporate cooperative network effects such as feedback inhibition, allosteric activation, and metabolite sharing. When a particular enzyme’s parameters would drive the system away from the target flux profile, neighboring enzymes are adjusted in a coordinated manner, reflecting the biological reality that metabolic pathways self‑regulate to maintain homeostasis.
To demonstrate the method, the authors focus on the central metabolism of M. extorquens AM1, a methylotrophic bacterium that oxidizes methanol to formaldehyde and assimilates carbon via the serine cycle, the ethylmalonyl‑CoA pathway, and a truncated TCA cycle. The model comprises 45 enzymatic steps, encompassing methanol dehydrogenation, formaldehyde oxidation, C1 assimilation, and energy‑generating reactions. Using Monte‑Carlo sampling, the authors generated over 10,000 random parameter sets; after thermodynamic and physiological filtering, roughly 200 sets survived. From these, the best‑fitting set reproduced experimentally measured fluxes (methanol uptake, CO₂ evolution, ATP/ADP ratios) with a mean absolute error of less than 10%, a substantial improvement over previous kinetic approximations that lacked systematic parameter inference. Sensitivity analyses revealed that the model’s output is relatively insensitive to moderate variations in individual parameters, confirming the robustness assumption. Moreover, simulated perturbations (e.g., reduced oxygen availability or increased methanol concentration) yielded flux responses that matched observed growth rate changes, indicating that the cooperative adjustments embedded in the workflow capture essential regulatory behavior.
The significance of this work lies in its demonstration that a complete kinetic parameterization is not a prerequisite for constructing functional, predictive metabolic models at the genome‑scale. By integrating thermodynamic feasibility, physiological flux constraints, and network‑level cooperation, the authors dramatically shrink the feasible parameter space, making the problem tractable with existing computational resources. The approach is modular: additional data types (transcriptomics, proteomics, metabolomics) can be incorporated as further constraints, and the sampling‑filtering pipeline can be coupled with machine‑learning techniques to accelerate convergence toward optimal parameter sets. The authors also discuss limitations, such as the reliance on accurate thermodynamic data and the assumption that the underlying network topology is correct; they propose future extensions that include dynamic regulation of enzyme expression and post‑translational modifications.
In conclusion, this study provides a practical blueprint for kinetic modeling of global metabolic networks under realistic data scarcity. It bridges the gap between static flux analysis and fully dynamic kinetic simulations, offering a scalable solution that can be applied to other microbes, plant systems, or even human cell metabolism where comprehensive kinetic datasets are rarely available. The methodology promises to enhance our ability to predict metabolic responses to genetic or environmental perturbations, thereby supporting metabolic engineering, drug target identification, and fundamental studies of cellular physiology.
Comments & Academic Discussion
Loading comments...
Leave a Comment