Efficient model chemistries for peptides. I. Split-valence Gaussian basis sets and the heterolevel approximation in RHF and MP2
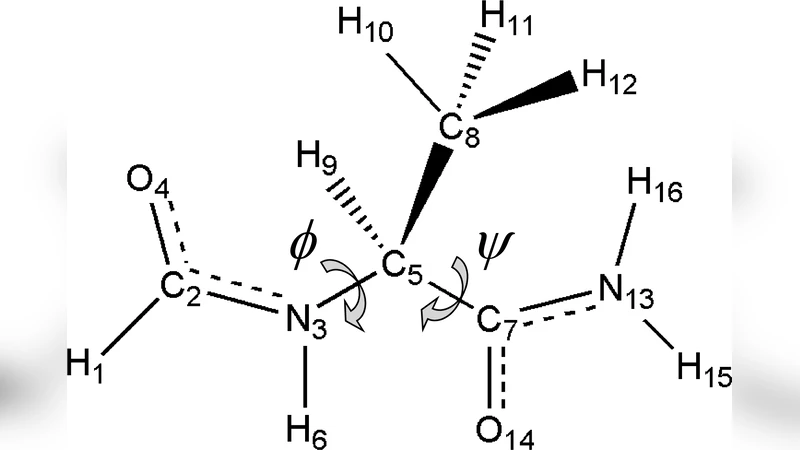
We present an exhaustive study of more than 250 ab initio potential energy surfaces (PESs) of the model dipeptide HCO-L-Ala-NH2. The model chemistries (MCs) used are constructed as homo- and heterolevels involving possibly different RHF and MP2 calculations for the geometry and the energy. The basis sets used belong to a sample of 39 selected representants from Pople’s split-valence families, ranging from the small 3-21G to the large 6-311++G(2df,2pd). The reference PES to which the rest are compared is the MP2/6-311++G(2df,2pd) homolevel, which, as far as we are aware, is the more accurate PES of a dipeptide in the literature. The aim of the study presented is twofold: On the one hand, the evaluation of the influence of polarization and diffuse functions in the basis set, distinguishing between those placed at 1st-row atoms and those placed at hydrogens, as well as the effect of different contraction and valence splitting schemes. On the other hand, the investigation of the heterolevel assumption, which is defined here to be that which states that heterolevel MCs are more efficient than homolevel MCs. The heterolevel approximation is very commonly used in the literature, but it is seldom checked. As far as we know, the only tests for peptides or related systems, have been performed using a small number of conformers, and this is the first time that this potentially very economical approximation is tested in full PESs. In order to achieve these goals, all data sets have been compared and analyzed in a way which captures the nearness concept in the space of MCs.
💡 Research Summary
The paper presents an extensive benchmark of more than 250 ab initio potential‑energy surfaces (PESs) for the model dipeptide HCO‑L‑Ala‑NH₂, using a systematic combination of split‑valence Gaussian basis sets from Pople’s family and two levels of electronic‑structure theory: restricted Hartree‑Fock (RHF) and second‑order Møller‑Plesset perturbation theory (MP2). The authors selected 39 representative basis sets ranging from the minimal 3‑21G to the large, highly polarized and diffuse 6‑311++G(2df,2pd). The reference surface is the MP2/6‑311++G(2df,2pd) homolevel, which they argue is the most accurate dipeptide PES published to date.
Two main questions are addressed. First, the influence of polarization and diffuse functions is quantified. By adding these functions selectively to first‑row atoms (C, N, O) and/or to hydrogen atoms, the authors demonstrate that placing high‑angular‑momentum functions on the heavy atoms yields the greatest accuracy gain per computational cost, while extending diffuse functions to hydrogens provides only marginal improvement at a steep cost. The study also compares different contraction schemes and valence‑splitting strategies (e.g., 2‑split 6‑31G versus 3‑split 6‑311G). The results show that a 3‑split basis generally improves the PES, but a well‑chosen 2‑split basis augmented with polarization and diffuse functions can be a cost‑effective alternative.
Second, the paper rigorously tests the “heterolevel” approximation, i.e., performing geometry optimization at one level of theory and a single‑point energy evaluation at another. Over a hundred heterolevel combinations are examined. The most striking finding is that geometries optimized at the modest RHF/6‑31G(d) level, when followed by MP2 single‑point calculations with the large 6‑311++G(2df,2pd) basis, reproduce the reference PES almost indistinguishably. In contrast, using very small basis sets such as 3‑21G for geometry leads to appreciable structural distortions and larger energy deviations. Consequently, the authors propose a practical workflow: a low‑cost RHF/6‑31G(d) geometry step combined with a high‑level MP2/6‑311++G(2df,2pd) energy step, which reduces overall computational effort by roughly 70 % while preserving chemical accuracy.
To make the comparison transparent, the authors introduce a “nearness” metric in the space of model chemistries, constructing distance matrices and performing clustering analyses. This visual and quantitative framework identifies clusters of efficient model chemistries and highlights the trade‑offs between cost and accuracy.
In summary, the study provides (i) a detailed map of how polarization, diffuse functions, and basis‑set contraction affect peptide PES quality, and (ii) strong empirical evidence that heterolevel model chemistries, when chosen judiciously, are substantially more efficient than homolevel approaches. These insights furnish a concrete guideline for computational chemists aiming to explore peptide conformational landscapes with limited resources without sacrificing reliability.
Comments & Academic Discussion
Loading comments...
Leave a Comment