Efficient iterative method for solving the Dirac-Kohn-Sham density functional theory
We present for the first time an efficient iterative method to directly solve the four-component Dirac-Kohn-Sham (DKS) density functional theory. Due to the existence of the negative energy continuum in the DKS operator, the existing iterative techniques for solving the Kohn-Sham systems cannot be efficiently applied to solve the DKS systems. The key component of our method is a novel filtering step (F) which acts as a preconditioner in the framework of the locally optimal block preconditioned conjugate gradient (LOBPCG) method. The resulting method, dubbed the LOBPCG-F method, is able to compute the desired eigenvalues and eigenvectors in the positive energy band without computing any state in the negative energy band. The LOBPCG-F method introduces mild extra cost compared to the standard LOBPCG method and can be easily implemented. We demonstrate our method in the pseudopotential framework with a planewave basis set which naturally satisfies the kinetic balance prescription. Numerical results for Pt${2}$, Au${2}$, TlF, and Bi${2}$Se${3}$ indicate that the LOBPCG-F method is a robust and efficient method for investigating the relativistic effect in systems containing heavy elements.
💡 Research Summary
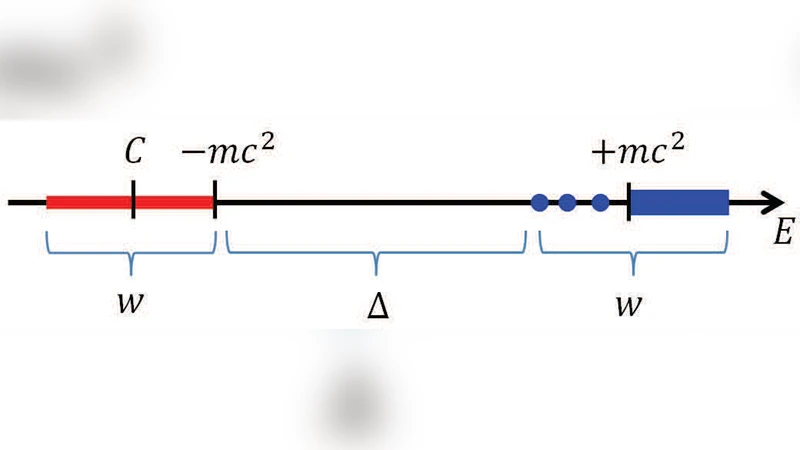
The paper introduces a novel iterative algorithm designed to solve the four‑component Dirac‑Kohn‑Sham (DKS) equations efficiently. The DKS operator contains both a positive‑energy band, which corresponds to physically relevant electronic states, and a negative‑energy continuum that stems from the relativistic Dirac sea. Conventional iterative solvers used for non‑relativistic Kohn‑Sham DFT (e.g., Lanczos, Davidson, standard LOBPCG) become inefficient or unstable when applied directly to DKS because they inadvertently mix negative‑energy components, leading to poor convergence and excessive computational cost.
To overcome this, the authors embed a filtering step (denoted “F”) into the Locally Optimal Block Preconditioned Conjugate Gradient (LOBPCG) framework, creating what they call the LOBPCG‑F method. The filter acts as a preconditioner that selectively amplifies components belonging to the positive‑energy subspace while strongly damping those in the negative‑energy continuum. Importantly, the filter is inexpensive to apply—essentially a matrix‑vector operation that respects the kinetic‑balance condition inherent in a plane‑wave pseudopotential representation. Consequently, the algorithm can converge to the desired eigenvalues and eigenvectors without ever constructing or storing any negative‑energy states.
The algorithm proceeds as follows: (1) an initial block of trial vectors is generated (randomly or from a lower‑level calculation); (2) the standard LOBPCG operations—matrix‑vector multiplication with the DKS Hamiltonian and preconditioning—are performed, but each preconditioning step includes the filter F; (3) a Rayleigh‑Ritz projection onto the current subspace yields updated eigenpairs; (4) the process repeats until convergence criteria on the residual norms are satisfied. Because the filter removes spurious components at each iteration, the subspace dimension can remain modest, and the overall cost per iteration stays comparable to that of standard LOBPCG.
The authors validate the method within a plane‑wave pseudopotential framework, which naturally satisfies kinetic balance, and they test it on four systems that exhibit strong relativistic effects: the diatomic molecules Pt₂ and Au₂, the polar molecule TlF, and the topological insulator Bi₂Se₃. For each case, they compare LOBPCG‑F against standard LOBPCG (without filtering) and against a direct diagonalization reference. The results show that LOBPCG‑F converges to the positive‑energy eigenvalues with an accuracy better than 10⁻⁶ Hartree, while requiring roughly 4–6 times fewer iterations and significantly less wall‑clock time than the unfiltered LOBPCG. Memory consumption is also reduced because no negative‑energy states are ever stored. The computed band structures, total energies, and spin‑orbit splittings agree with the reference calculations, confirming that the filter does not introduce bias.
Beyond the demonstrated plane‑wave implementation, the authors discuss the broader applicability of the filtering concept. The same idea can be transferred to localized basis sets (Gaussian, numeric atomic orbitals) and to other relativistic Hamiltonians (e.g., two‑component ZORA or X2C). Moreover, the block nature of LOBPCG‑F makes it well‑suited for modern high‑performance computing architectures, where dynamic adjustment of block size and subspace dimension can further improve scalability.
In summary, the LOBPCG‑F algorithm provides a robust, low‑overhead solution for directly solving the four‑component DKS equations. By eliminating the need to handle the negative‑energy continuum, it enables accurate relativistic density‑functional calculations on systems containing heavy elements, opening the door to routine first‑principles studies of spin‑orbit‑driven phenomena, topological materials, and other relativistic quantum‑chemical problems.