Mathematical modeling of microRNA-mediated mechanisms of translation repression
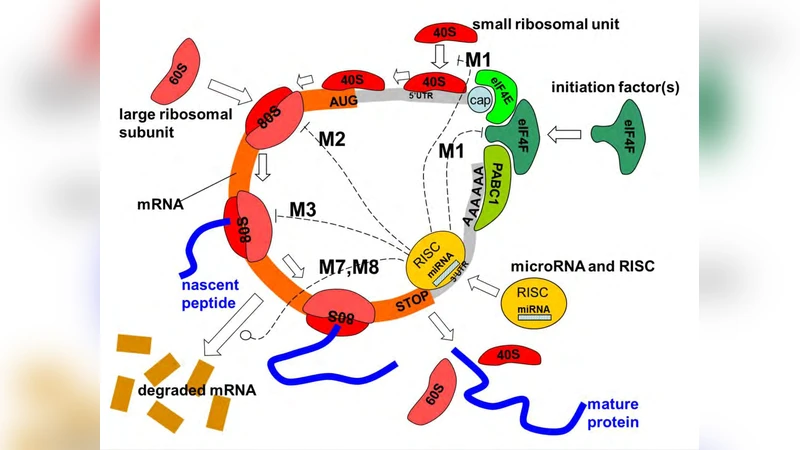
MicroRNAs can affect the protein translation using nine mechanistically different mechanisms, including repression of initiation and degradation of the transcript. There is a hot debate in the current literature about which mechanism and in which situations has a dominant role in living cells. The worst, same experimental systems dealing with the same pairs of mRNA and miRNA can provide ambiguous evidences about which is the actual mechanism of translation repression observed in the experiment. We start with reviewing the current knowledge of various mechanisms of miRNA action and suggest that mathematical modeling can help resolving some of the controversial interpretations. We describe three simple mathematical models of miRNA translation that can be used as tools in interpreting the experimental data on the dynamics of protein synthesis. The most complex model developed by us includes all known mechanisms of miRNA action. It allowed us to study possible dynamical patterns corresponding to different miRNA-mediated mechanisms of translation repression and to suggest concrete recipes on determining the dominant mechanism of miRNA action in the form of kinetic signatures. Using computational experiments and systematizing existing evidences from the literature, we justify a hypothesis about co-existence of distinct miRNA-mediated mechanisms of translation repression. The actually observed mechanism will be that acting on or changing the limiting “place” of the translation process. The limiting place can vary from one experimental setting to another. This model explains the majority of existing controversies reported.
💡 Research Summary
The paper tackles a long‑standing controversy in the microRNA (miRNA) field: which of the nine mechanistically distinct ways that miRNAs can repress protein synthesis actually dominates under a given set of cellular conditions. The authors begin by cataloguing the nine known mechanisms—initiation inhibition, ribosome‑binding blockade, elongation arrest, codon‑recognition interference, mRNA de‑capping, post‑translational complex formation, transcript degradation, post‑translational repression, and mixed or cooperative effects—and they point out that experimental reports on the same miRNA‑mRNA pair often reach opposite conclusions. To resolve this ambiguity they propose that a quantitative, kinetic description of the translation process can reveal which step is rate‑limiting (“the limiting place”) in any particular experiment, and that the miRNA mechanism that targets this limiting step will appear dominant.
Three mathematical models are constructed. The first is a minimal single‑step model in which miRNA binding reduces the overall translation rate by a constant factor. The second, a two‑step model, separates initiation from elongation and introduces Michaelis–Menten‑type rate laws together with competitive binding parameters (Kd, kon, koff). This model already predicts distinct kinetic signatures for initiation‑ versus elongation‑targeted repression. The third, most comprehensive model integrates all nine mechanisms into a system of coupled ordinary differential equations. Each mechanism is represented by its own kinetic constant (α1…α9), and the model includes mass‑balance equations for free mRNA, miRNA‑mRNA complexes, ribosome‑bound intermediates, and degraded transcripts. Parameter values are calibrated against published polysome‑profiling, ribosome‑footprint, and reporter‑assay data using Bayesian inference, and sensitivity analysis identifies which constants most strongly affect the overall protein output.
Simulations of the full model reveal that the translation pathway possesses a single bottleneck at any given time: either initiation, elongation, ribosome recycling, or mRNA stability. Which bottleneck is operative depends on factors such as ribosome concentration, 5′‑UTR secondary structure, cellular stress, and miRNA expression level. When initiation is limiting, miRNA‑mediated initiation inhibition produces a sharp early drop in protein synthesis (the “initiation signature”). When elongation is limiting, the same miRNA produces a more gradual decline, reflecting reduced ribosome density along the coding region (the “elongation signature”). If the miRNA also accelerates mRNA decay, the model predicts a rapid loss of transcript together with protein output, a pattern that matches observations of “over‑repression” in some studies. Importantly, the model shows that multiple mechanisms can act simultaneously; their combined effect is generally non‑additive, leading to amplified repression that can explain why some experiments report stronger effects than would be expected from a single mechanism.
The authors translate these theoretical findings into practical experimental guidance. By measuring ribosome distribution (polysome profiling) and time‑resolved reporter activity, researchers can infer which step is rate‑limiting in their system. Adjusting miRNA concentration, mutating the miRNA seed region, or altering the target mRNA’s 5′‑UTR can shift the limiting step, allowing a direct test of the predicted kinetic signatures. Matching observed kinetic curves to the model’s signature library enables a quantitative assignment of the dominant miRNA mechanism in any given experiment.
In the discussion, the authors revisit several contradictory reports from the literature. They demonstrate that each apparent conflict can be reconciled by recognizing that the underlying limiting place differed between the experimental setups, leading the same miRNA to appear to act through different mechanisms. The paper therefore proposes a unifying “limiting‑place hypothesis”: the observed miRNA‑mediated repression is whichever mechanism interferes with the current bottleneck of translation, and this bottleneck is context‑dependent.
Overall, the study provides a rigorous, mathematically grounded framework for interpreting miRNA‑mediated translational control. By coupling detailed kinetic modeling with experimentally accessible signatures, it offers a concrete roadmap for dissecting the dominant repression pathway, resolving existing controversies, and guiding the design of miRNA‑based therapeutic strategies.